Running command-line tools with shell scripts
Week 4 - shell scripts - part II
Overview and setting up
This session focuses on using programs, like various bioinformatics tools, with command-line interfaces (CLIs), and on running these inside shell scripts.
The strategy that you’ll learn is to write scripts that run a single program a single time, even if you need to run the program many times: in that case, you’ll loop over files outside of that script.
This means you’ll also need a higher-level “runner” script in which you save the looping code and more. We will talk about why this strategy makes sense, especially when you have a supercomputer at your disposal.
We’ll start with a quick intro to FASTQ files and the FastQC program, which will be our first example of a CLI tool.
Our practice data set
Our practice data set is from the paper “Two avian Plasmodium species trigger different transcriptional responses on their vector Culex pipiens”, published last year in Molecular Ecology (link):

This paper uses RNA-seq data to study gene expression in Culex pipiens mosquitos infected with malaria-causing Plasmodium protozoans — specifically, it compares mosquitos according to:
- Infection status: Plasmodium cathemerium vs. P. relictum vs. control
- Time after infection: 24 h vs. 10 days vs. 21 days
However, to keep things manageable for our practice, I have subset the data to omit the 21-day samples and only keep 500,000 reads per FASTQ file. All in all, our set of files consists of:
- 44 paired-end Illumina FASTQ files for 22 samples.
- Culex pipiens reference genome file from NCBI: assembly in FASTA format and annotation in GTF format.
- A metadata file in TSV format with sample IDs and treatment & time point info.
- A README file describing the data set.
Get your own copy of the data
# (Assuming you are in /fs/ess/PAS2700/users/$USER)
cd week04
cp -rv /fs/ess/PAS2700/share/garrigos_data .‘/fs/ess/PAS2700/share/garrigos_data’ -> ‘./garrigos_data’
‘/fs/ess/PAS2700/share/garrigos_data/meta’ -> ‘./garrigos_data/meta’
‘/fs/ess/PAS2700/share/garrigos_data/meta/metadata.tsv’ -> ‘./garrigos_data/meta/metadata.tsv’
‘/fs/ess/PAS2700/share/garrigos_data/ref’ -> ‘./garrigos_data/ref’
‘/fs/ess/PAS2700/share/garrigos_data/ref/GCF_016801865.2.gtf’ -> ‘./garrigos_data/ref/GCF_016801865.2.gtf’
‘/fs/ess/PAS2700/share/garrigos_data/ref/GCF_016801865.2.fna’ -> ‘./garrigos_data/ref/GCF_016801865.2.fna’
‘/fs/ess/PAS2700/share/garrigos_data/fastq’ -> ‘./garrigos_data/fastq’
‘/fs/ess/PAS2700/share/garrigos_data/fastq/ERR10802868_R2.fastq.gz’ -> ‘./garrigos_data/fastq/ERR10802868_R2.fastq.gz’
‘/fs/ess/PAS2700/share/garrigos_data/fastq/ERR10802863_R1.fastq.gz’ -> ‘./garrigos_data/fastq/ERR10802863_R1.fastq.gz’
‘/fs/ess/PAS2700/share/garrigos_data/fastq/ERR10802880_R2.fastq.gz’ -> ‘./garrigos_data/fastq/ERR10802880_R2.fastq.gz’
‘/fs/ess/PAS2700/share/garrigos_data/fastq/ERR10802880_R1.fastq.gz’ -> ‘./garrigos_data/fastq/ERR10802880_R1.fastq.gz’
‘/fs/ess/PAS2700/share/garrigos_data/fastq/ERR10802870_R1.fastq.gz’ -> ‘./garrigos_data/fastq/ERR10802870_R1.fastq.gz’
# [...output truncated...]Take a look at the files you just copied with the tree command, a sort of recursive ls with simple tree-like output:
# -C will add colors (not shown in the output below)
tree -C garrigos_datagarrigos_data
├── fastq
│ ├── ERR10802863_R1.fastq.gz
│ ├── ERR10802863_R2.fastq.gz
│ ├── ERR10802864_R1.fastq.gz
│ ├── ERR10802864_R2.fastq.gz
│ ├── [...other FASTQ files not shown...]
├── meta
│ └── metadata.tsv
├── README.md
└── ref
├── GCF_016801865.2.fna
└── GCF_016801865.2.gtf
3 directories, 48 files1 FASTQ files and FastQC
1.1 The FASTQ format
FASTQ is a very common output format of high-throughput sequencing machines. Like most genomic data files, these are plain text files. Each sequence that is read by the sequencer (i.e., each “read”) forms one FASTQ entry represented by four lines. The lines contain, respectively:
- A header that starts with
@and e.g. uniquely identifies the read - The nucleotide sequence itself
- A
+(plus sign) - One-character quality scores for each base in the sequence
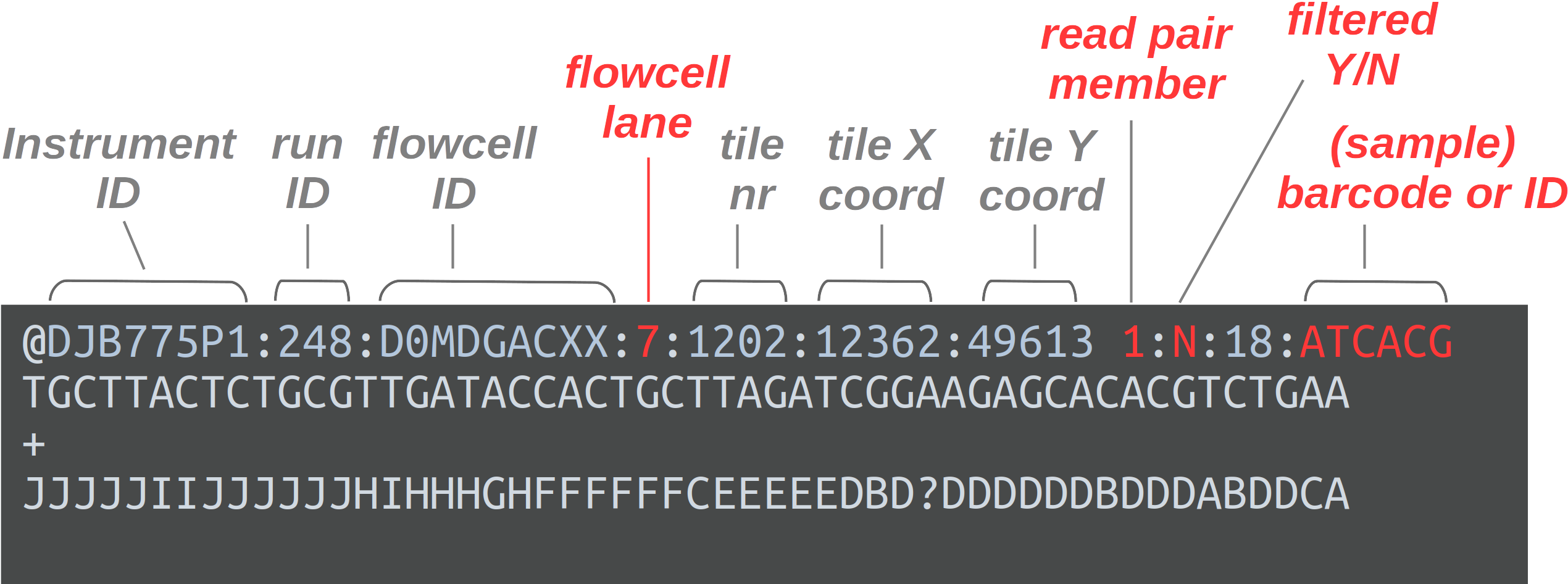
The header line is annotated, with some of the more useful components highlighted in red.
For viewing purposes, this read (at only 56 bp) is shorter than what is typical.
The “Q” in FASTQ stands for “quality”, to contrast this format with FASTA, a more basic and generic sequence data format that does not include base quality scores. FASTQ files have the extension .fastq or .fq, but they are very commonly gzip-compressed, in which case their name ends in .fastq.gz or .fq.gz.
The quality scores we saw in the read above represent an estimate of the error probability of the base call. Specifically, they correspond to a numeric “Phred” quality score (Q), which is a function of the estimated probability that a base call is erroneous (P):
Q = -10 * log10(P)
For some specific probabilities and their rough qualitative interpretations for Illumina data:
| Phred quality score | Error probability | Rough interpretation | ASCII character |
|---|---|---|---|
| 10 | 1 in 10 | terrible | + |
| 20 | 1 in 100 | bad | 5 |
| 30 | 1 in 1,000 | good | ? |
| 40 | 1 in 10,000 | excellent | ? |
This numeric quality score is represented in FASTQ files not by the number itself, but by a corresponding “ASCII character” (last column in the table). This allows for a single-character representation of each possible score — as a consequence, each quality score character can conveniently correspond to (& line up with) a base character in the read. (For your reference, here is a complete lookup table — look at the top table, “BASE=33”).
1.2 Our FASTQ files
Take a look at a file listing of your FASTQ files:
ls -lh garrigos_data/fastqtotal 941M
-rw-r-----+ 1 jelmer PAS0471 21M Mar 21 09:38 ERR10802863_R1.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 22M Mar 21 09:38 ERR10802863_R2.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 21M Mar 21 09:38 ERR10802864_R1.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 22M Mar 21 09:38 ERR10802864_R2.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 22M Mar 21 09:38 ERR10802865_R1.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 22M Mar 21 09:38 ERR10802865_R2.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 21M Mar 21 09:38 ERR10802866_R1.fastq.gz
# [...output truncated...]Note that:
- There are two files per sample:
_R1(forward reads) and_R2(reverse reads). - The files all have a
.gzextension, indicating they have been compressed with the gzip utility. - The files are ~21-22 Mb in size — considerably smaller than the original file sizes (around 1-2 Gb, which is typical) because they were subsampled.
1.3 Viewing the contents of FASTQ files
Next, try to take a peak inside one of these FASTQ files. Use -n 8 with head to print the first 8 lines (2 reads):
head -n 8 garrigos_data/fastq/ERR10802863_R1.fastq.gz�
Խے�8�E��_1f�"�QD�J��D�fs{����Yk����d��*��
|��x���l�j�N������?������ٔ�bUs�Ng�Ǭ���i;_��������������|<�v����3��������|���ۧ��3ĐHyƕ�bIΟD�%����Sr#~��7��ν��1y�Ai,4
w\]"b�#Q����8��+[e�3d�4H���̒�l�9LVMX��U*�M����_?���\["��7�s\<_���:�$���N��v�}^����sw�|�n;<�<�oP����
i��k��q�ְ(G�ϫ��L�^��=��<���K��j�_/�[ۭV�ns:��U��G�z�ݎ�j����&��~�F��٤ZN�'��r2z}�f\#��:�9$�����H�݂�"�@M����H�C�
�0�pp���1�O��I�H�P됄�.Ȣe��Q�>���
�'�;@D8���#��St�7k�g��|�A䉻���_���d�_c������a\�|�_�mn�]�9N������l�٢ZN�c�9u�����n��n�`��
"gͺ�
���H�?2@�FC�S$n���Ԓh� nԙj��望��f �?N@�CzUlT�&�h�Pt!�r|��9~)���e�A�77�h{��~�� ��
# [...output truncated...]Ouch! 😳 What went wrong here? (Click for the solution)
We were directly presented with the contents of the compressed file, which isn’t human-readable.To get around the problem we just encountered with head, we might be inclined to uncompress these files, which we could do with the gunzip command. However, uncompressed files take up several times as much disk storage space as compressed ones. Fortunately, we don’t need to decompress them:
- Almost any bioinformatics tool will accept compressed FASTQ files.
- We can still view these files in compressed form, as shown below.
Instead, we’ll use the less command, which automatically displays gzip-compressed files in human-readable form:
less -S garrigos_data/fastq/ERR10802863_R1.fastq.gz@ERR10802863.8435456 8435456 length=74
CAACGAATACATCATGTTTGCGAAACTACTCCTCCTCGCCTTGGTGGGGATCAGTACTGCGTACCAGTATGAGT
+
AAAAAEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEE
@ERR10802863.27637245 27637245 length=74
GCCACACTTTTGAAGAACAGCGTCATTGTTCTTAATTTTGTCGGCAACGCCTGCACGAGCCTTCCACGTAAGTT
+
AAAAAEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEAEE<EEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEE
@ERR10802863.10009244 10009244 length=73
CTCGGCGTTAACTTCATCACGCAGATCATTCCGTTCCAGCAGCTGAAGCAAGACTACCGTCAGTACGAGATGA
+
AAAAAEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEE
@ERR10802863.6604176 6604176 length=74
AACTACAAATCTTCCTGTGCCGTTTCCAGCAAGTACGTCGATACCTTCGATGGACGCAACTACGAGTACAACAT
+
AAAAAEAEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEE
@ERR10802863.11918285 11918285 length=35
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+
###################################-S option to less suppresses line-wrapping: lines in the file will not be “wrapped” across multiple lines.
Exercise: Explore the file with less
After running the command above, you should be viewing the file inside the less pager.
You can move around in the file in several ways: by scrolling with your mouse, with up and down arrows, or, if you have them, PgUp and PgDn keys (also, u will move up half a page and d down half a page).
Recall that you won’t get your shell prompt back until you press q to quit less.
Do you notice anything that strikes you as potentially unusual, some reads that look different from others?
Click for the solution
There are a number of reads that are much shorter than the others and only consist of N, i.e. uncalled bases. For example:
@ERR10802863.11918285 11918285 length=35
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+
###################################1.4 FastQC
FastQC is a ubiquitous tool for quality control of FASTQ files. Running FastQC or a similar program is the first step in nearly any high-throughput sequencing project. FastQC is also a good introductory example of a tool with a command-line interface.
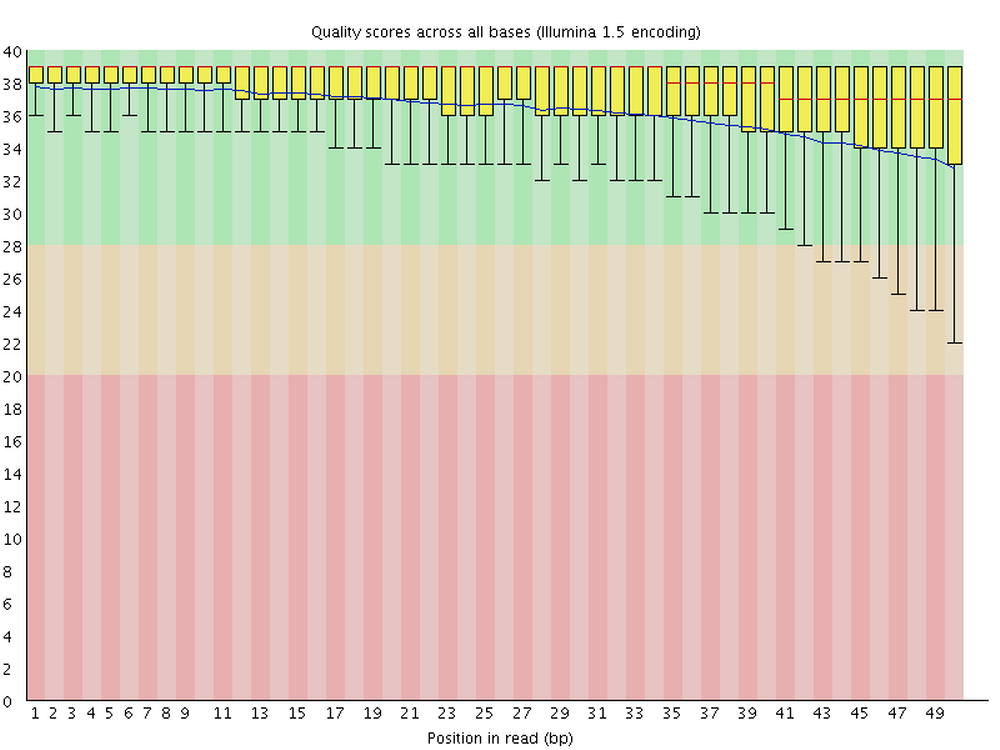
For each FASTQ file, FastQC outputs an HTML file that you can open in your browser with about a dozen graphs showing different QC metrics. The most important one is the per-base quality score graph:
2 Running FastQC interactively
The command fastqc will run the FastQC program. If you want to analyze a FASTQ file with default FastQC settings, a complete FastQC command would simply be fastqc followed by the file name:
# (Don't run this)
fastqc garrigos_data/fastq/ERR10802863_R1.fastq.gzHowever, an annoying FastQC default behavior is that it writes its output files in the same dir that contains the input FASTQ files — this means mixing your raw data with your results, which we don’t want!
To figure out how we can change that behavior, first consider that many commands and bioinformatics tools alike have an option -h and/or --help to print usage information to the screen. Let’s try that:
fastqc -hbash: fastqc: command not found...However, there is a wrinkle — while FastQC is installed at OSC1, we have to first “load it”2:
module load fastqc/0.11.8Exercise: FastQC help and output dir
Print FastQC’s help info, and figure out which option you can use to specify a custom output directory.
Click for the solution
Running fastqc -h or fastqc --help will work to show the help info. You’ll get quite a bit of output printed to screen, including the snippet about output directories that is reproduced below:
fastqc -h -o --outdir Create all output files in the specified output directory.
Please note that this directory must exist as the program
will not create it. If this option is not set then the
output file for each sequence file is created in the same
directory as the sequence file which was processed.-o or equivalently, --outdir to specify an output dir.
With the added --outdir (or -o) option, let’s try to run the following FastQC command:
# We'll have to first create the outdir ourselves, in this case
mkdir -p results/fastqc
# Now we run FastQC
fastqc --outdir results/fastqc garrigos_data/fastq/ERR10802863_R1.fastq.gzStarted analysis of ERR10802863_R1.fastq.gz
Approx 5% complete for ERR10802863_R1.fastq.gz
Approx 10% complete for ERR10802863_R1.fastq.gz
Approx 15% complete for ERR10802863_R1.fastq.gz
[...truncated...]
Analysis complete for ERR10802863_R1.fastq.gzIn the output dir we specified, we have a .zip file, which contains tables with FastQC’s data summaries, and an .html (HTML) file, which contains the graphs:
ls -lh results/fastqctotal 512K
-rw-rw----+ 1 jelmer PAS0471 241K Mar 21 09:53 ERR10802863_R1_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 256K Mar 21 09:53 ERR10802863_R1_fastqc.zipFastQC allows us to specify the output directory, but not the output file names: these will be automatically determined based on the input file name(s). This kind of behavior is fairly common for bioinformatics programs, since they will often produce multiple output files.
Exercise: Another FastQC run
Run FastQC for the corresponding R2 FASTQ file. Would you use the same output dir or a separate one?
Click for the solution
Yes, it makes sense to use the same output dir, since as you could see above, the output file names have the input file identifiers in them. As such, we don’t need to worry about overwriting files, and it will be more convenient to have all results in a single dir.
To run FastQC for the R2 (reverse-read) file:
fastqc --outdir results/fastqc garrigos_data/fastq/ERR10802863_R2.fastq.gzStarted analysis of ERR10802863_R2.fastq.gz
Approx 5% complete for ERR10802863_R2.fastq.gz
Approx 10% complete for ERR10802863_R2.fastq.gz
Approx 15% complete for ERR10802863_R2.fastq.gz
[...truncated...]
Analysis complete for ERR10802863_R2.fastq.gzls -lh results/fastqctotal 1008K
-rw-rw----+ 1 jelmer PAS0471 241K Mar 21 09:53 ERR10802863_R1_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 256K Mar 21 09:53 ERR10802863_R1_fastqc.zip
-rw-rw----+ 1 jelmer PAS0471 234K Mar 21 09:55 ERR10802863_R2_fastqc.html
-rw-rw----+ 1 jelmer PAS0471 244K Mar 21 09:55 ERR10802863_R2_fastqc.zipNow, we have four files: two for each of our preceding successful FastQC runs.
3 Running FastQC with a shell script
Instead of running FastQC interactively, we’ll want to write a script that runs it. Specifically, our script will deliberately run FastQC on only one FASTQ file.
In bioinformatics, you commonly need to run a CLI tool many times, because most tools can or have to be run separately run for each file or sample. Instead of writing a script that runs one file or sample, a perhaps more intuitive approach would be writing a script that processes all files/samples in a single run. That can be accomplished by:
- Looping over files/samples inside the script; or
- Passing many file names or a glob with
*to a single run of the tool (this can be done with some tools).
However, given that we have access to OSC’s clusters, it will save running time -potentially a lot of it- when we submit a separate batch job for each FASTQ file. This is why we will write a script such that runs only one file, and then we’ll run that script many times using a loop outside of te script.
For now, we’ll practice with writing scripts this way, and running them interactively. Next week, we will take the next step and submit each run of the script as a batch job.
3.1 Arguments to the script
Our favored approach of running the script for one FASTQ file at a time means that our script needs to accept a FASTQ file name as an argument. So instead of using a line like the one we ran above…
# [Don't copy or run this]
fastqc --outdir results/fastqc garrigos_data/fastq/ERR10802863_R2.fastq.gz…we would use a variable for the file name — for example:
# [Don't copy or run this]
fastqc --outdir results/fastqc "$fastq_file"And while we’re at it, we may also want to use a variable for the output dir:
# [Don't copy or run this]
fastqc --outdir "$outdir" "$fastq_file"Of course, these variables don’t appear out of thin air completely — we need to pass arguments to the script, and copy the placeholder variables inside the script:
# [Don't copy or run this]
# Copy the placeholder variables
fastq_file=$1
outdir=$2
# Run FastQC
fastqc --outdir "$outdir" "$fastq_file"And such a script would be run for a single file as follows:
# [Don't copy or run this]
# Syntax: 'bash <script-path> <argument1> <argument2>'
bash scripts/fastqc.sh garrigos_data/fastq/ERR10802863_R2.fastq.gz results/fastqcAnd it would be run for all files by looping over all them as follows:
# [Don't copy or run this]
# Run the script separately for each FASTQ file
for fastq_file in data/fastq/*fastq.gz; do
bash scripts/fastqc.sh "$fastq_file" results/fastqc
done3.2 Creating an initial script
We saw some code to run FastQC inside a script, to which we should add a number of “boilerplate” bits of code:
The shebang line and strict Bash settings:
#!/bin/bash set -euo pipefailA line to load the relevant OSC software module:
module load fastqc/0.11.8A line to create the output directory if it doesn’t yet exist:
mkdir -p "$outdir"
-p option to mkdir (Click to expand)
Using the -p option does two things at once, and both are necessary for a foolproof inclusion of this command in a script:
It will enable
mkdirto create multiple levels of directories at once (i.e., to act recursively): by default,mkdirerrors out if the parent directory/ies of the specified directory don’t yet exist.mkdir newdir1/newdir2mkdir: cannot create directory ‘newdir1/newdir2’: No such file or directory# This successfully creates both directories mkdir -p newdir1/newdir2If the directory already exists, it won’t do anything and won’t return an error. Without this option,
mkdirwould return an error in this case, which would in turn lead the script to abort at that point with oursetsettings:mkdir newdir1/newdir2mkdir: cannot create directory ‘newdir1/newdir2’: File exists# This does nothing since the dirs already exist mkdir -p newdir1/newdir2
With those additions, our partial script would look like this:
# [Don't copy or run this - we'll add to it later]
#!/bin/bash
set -euo pipefail
# Load the OSC module for FastQC
module load fastqc
# Copy the placeholder variables
fastq_file=$1
outdir=$2
# Create the output dir if needed
mkdir -p "$outdir"
# Run FastQC
fastqc --outdir "$outdir" "$fastq_file"Notice that this script to run a CLI tool is very similar to our “toy scripts” from the previous sessions: mostly standard (“boilerplate”) code with just a single command to run our program of interest. Therefore, you can adopt this script as a template for scripts that run other command-line programs, and will generally only need minor modifications!
3.3 Add some “logging” statements
It is often useful to have your scripts “report” or “log” what is going on. For instance:
- At what date and time did we run this script.
- Which arguments were passed to the script.
- What are the designated output dirs/files.
- Perhaps even summaries of the output (we won’t do this here).
All of this can help with troubleshooting and record-keeping3. Let’s try this with our FastQC script:
#!/bin/bash
set -euo pipefail
# Load the OSC module for FastQC
module load fastqc
# Copy the placeholder variables
fastq_file=$1
outdir=$2
# Initial reporting
echo "# Starting script fastqc.sh"
date
echo "# Input FASTQ file: $fastq_file"
echo "# Output dir: $outdir"
echo
# Create the output dir if needed
mkdir -p "$outdir"
# Run FastQC
fastqc --outdir "$outdir" "$fastq_file"
# Final reporting
echo
echo "# Done with script fastqc.sh"
dateA couple of notes about the lines that were added to the script above:
- We printed a “marker line”
Done with scriptthat indicates the end of the script was reached. This is handy due to oursetsettings: seeing this line printed means that no errors were encountered. - Running
dateat the beginning and end of the script is one way to check the running time of the script. - Printing the input files can be particularly useful for troubleshooting.
- The lines that only have
echowill simply print a blank line, basically as a separator between sections.
Create a script to run FastQC:
touch scripts/fastqc.shOpen it and paste in the code in the box above.
4 A “runner” script
4.1 Running our FastQC script for 1 file
Let’s run our FastQC script for one FASTQ file:
bash scripts/fastqc.sh garrigos_data/fastq/ERR10802863_R1.fastq.gz results/fastqc# Starting script fastqc.sh
Wed Mar 27 21:53:13 EDT 2024
# Input FASTQ file: garrigos_data/fastq/ERR10802863_R1.fastq.gz
# Output dir: results/fastqc
Started analysis of ERR10802863_R1.fastq.gz
Approx 5% complete for ERR10802863_R1.fastq.gz
Approx 10% complete for ERR10802863_R1.fastq.gz
Approx 15% complete for ERR10802863_R1.fastq.gz
Approx 20% complete for ERR10802863_R1.fastq.gz
Approx 25% complete for ERR10802863_R1.fastq.gz
Approx 30% complete for ERR10802863_R1.fastq.gz
Approx 35% complete for ERR10802863_R1.fastq.gz
Approx 40% complete for ERR10802863_R1.fastq.gz
Approx 45% complete for ERR10802863_R1.fastq.gz
Approx 50% complete for ERR10802863_R1.fastq.gz
Approx 55% complete for ERR10802863_R1.fastq.gz
Approx 60% complete for ERR10802863_R1.fastq.gz
Approx 65% complete for ERR10802863_R1.fastq.gz
Approx 70% complete for ERR10802863_R1.fastq.gz
Approx 75% complete for ERR10802863_R1.fastq.gz
Approx 80% complete for ERR10802863_R1.fastq.gz
Approx 85% complete for ERR10802863_R1.fastq.gz
Approx 90% complete for ERR10802863_R1.fastq.gz
Approx 95% complete for ERR10802863_R1.fastq.gz
Approx 100% complete for ERR10802863_R1.fastq.gz
Analysis complete for ERR10802863_R1.fastq.gz
# Done with script fastqc.sh
Wed Mar 27 21:53:19 EDT 2024However, as discussed above, we’ll want to run the script for each FASTQ file. We’ll have to write a loop to do so, and that loop will go in a “runner script”.
4.2 What are runner scripts and why do we need them
The loop code could be directly typed in the terminal, but it’s better to save this in a file/script as well.
We will now create such a file, which has the overall purpose of documenting the steps we took. You can think of this file as akin to an analysis lab notebook4. Because it will contain shell code, we will save it as a shell script (.sh) just like the script to run fastqc.sh and other individual analysis steps.
However, it is important to realize that this script is conceptually different from the scripts that run individual steps of your analysis. The latter are meant to be run/submitted in their entirety by the runner script, whereas commands in the former typically have to be run one-by-one, i.e. interactively. This kind of script is sometimes called a “runner” or “master” script.
To summarize, we’ll separate our code into two hierarchical levels of scripts:
- Scripts that run individual steps of your analysis, like
fastqc.sh. These will eventually be submitted a batch jobs. - An overarching “runner” script with code that we run interactively, to orchestrates batch job submission of the individual steps.
To make this division clearer, we’ll also save these scripts in separate directories:
scriptsfor the analysis scripts.runfor the runner script(s).
It is a good idea to keep the shell scripts you will submit (e.g., fastqc.sh) simple in the sense that they should generally just run one program, and not a sequence of programs.
Once you get the hang of writing these scripts, it may seem appealing to string a series of programs/steps together in a single script, so that it’s easier to rerun everything at once — but in practice, that will often end up leading to more difficulties than convenience. If you do want to develop a workflow that can be easily run and rerun from start to finish, you should learn a workflow management system like Snakemake or Nextflow — we will talk about Nextflow in week 6.
First off, not that this applies only once we start submitting our scripts as batch jobs.
Once we’ve added multiple batch job steps, and the input of a later step uses the output of an earlier step, we won’t be able to just run the script as is. This is because the runner script would then submit jobs from different steps all at once, and that later step would start running before the earlier step has finished.
For example, consider the following series of two steps, in which the second step uses the output of the first step:
# This script would create a genome "index" for STAR, that will be used in the next step
# ('my_genome.fa' = input genome FASTA, 'results/star_index' = output index dir)
sbatch scripts/star_index.sh my_genome.fa results/star_index
# This script would align a FASTQ file to the genome index created in the previous step
# ('results/star_index' = input index dir, 'sampleA.fastq.gz' = input FASTQ file,
# 'results/star_align' = output dir)
sbatch scripts/star_align.sh results/star_index sampleA.fastq.gz results/star_align If these two lines were included in your runner script, and you would run that script in its entirety all at once, the script in the second step would be submitted just a split-second after the first one (when using sbatch, you get your prompt back immediately – there is no waiting). As such, it would fail because of the missing output from the first step.
It is possible to make sbatch batch jobs wait for earlier steps to finish (e.g. with the --dependency option), but this quickly gets tricky. If you want to create a workflow/pipeline that can run from start to finish in an automated way, you should consider using a workflow management system like Snakemake or NextFlow — we will talk about Nextflow in week 6!
4.3 Creating our runner script
Create a new file, and open it after running these commands:
mkdir run
touch run/run.shIn this script, add the code that runs our fastqc.sh script for each FASTQ file, and then run that code:
# Run FastQC for each FASTQ file
for fastq_file in garrigos_data/fastq/*fastq.gz; do
bash scripts/fastqc.sh "$fastq_file" results/fastqc
done# Starting script fastqc.sh
Thu Mar 21 10:06:46 EDT 2024
# Input FASTQ file: garrigos_data/fastq/ERR10802863_R1.fastq.gz
# Output dir: results/fastqc
Started analysis of ERR10802863_R1.fastq.gz
Approx 5% complete for ERR10802863_R1.fastq.gz
Approx 10% complete for ERR10802863_R1.fastq.gz
Approx 15% complete for ERR10802863_R1.fastq.gz
Approx 20% complete for ERR10802863_R1.fastq.gz
Approx 25% complete for ERR10802863_R1.fastq.gz
Approx 30% complete for ERR10802863_R1.fastq.gz
Approx 35% complete for ERR10802863_R1.fastq.gz
Approx 40% complete for ERR10802863_R1.fastq.gz
Approx 45% complete for ERR10802863_R1.fastq.gz
Approx 50% complete for ERR10802863_R1.fastq.gz
Approx 55% complete for ERR10802863_R1.fastq.gz
Approx 60% complete for ERR10802863_R1.fastq.gz
Approx 65% complete for ERR10802863_R1.fastq.gz
Approx 70% complete for ERR10802863_R1.fastq.gz
Approx 75% complete for ERR10802863_R1.fastq.gz
Approx 80% complete for ERR10802863_R1.fastq.gz
Approx 85% complete for ERR10802863_R1.fastq.gz
Approx 90% complete for ERR10802863_R1.fastq.gz
Approx 95% complete for ERR10802863_R1.fastq.gz
Approx 100% complete for ERR10802863_R1.fastq.gz
Analysis complete for ERR10802863_R1.fastq.gz
# Done with script fastqc.sh
Thu Mar 21 10:06:51 EDT 2024
# Starting script fastqc.sh
Thu Mar 21 10:06:51 EDT 2024
# Input FASTQ file: garrigos_data/fastq/ERR10802863_R2.fastq.gz
# Output dir: results/fastqc
Started analysis of ERR10802863_R2.fastq.gz
Approx 5% complete for ERR10802863_R2.fastq.gz
Approx 10% complete for ERR10802863_R2.fastq.gz
# [...output truncated...]5 Looping over samples rather than files
In some cases, we can’t simply loop over all files like we have done so far. For example, in many tools that process paired-end FASTQ files, the corresponding R1 and R2 files for each sample must be processed together. That is, we don’t run the tool separately for each FASTQ file, but separately for each sample i.e. each pair of FASTQ files.
How can we loop over pairs of FASTQ files instead? There are two main ways:
- Create a list of sample IDs, loop over these IDs, and find the pair of FASTQ files with matching names.
- Loop over the R1 files only and then infer the name of the corresponding R2 file within the loop. This is generally straightforward because the file names should be identical other than the read-direction identifier (
R1/R2).
Below, we will use the second method — but first, we’ll recap/learn a few prerequisites.
5.1 Recap of basename, and dirname
Running the basename command on a filename will strip any directories in its name:
basename garrigos_data/fastq/ERR10802863_R1.fastq.gzERR10802863_R1.fastq.gzYou can also provide any arbitrary suffix to also strip from the file name:
basename garrigos_data/fastq/ERR10802863_R1.fastq.gz .fastq.gzERR10802863_R1If you instead want the directory part of the path, use the dirname command:
dirname garrigos_data/fastq/ERR10802863_R1.fastq.gzgarrigos_data/fastq5.2 Parameter expansion
You can use so-called “parameter expansion”, with parameter basically being another word for variable, to search-and-replace text in your variable’s values. For example:
Assign a short DNA sequence to a variable:
dna_seq="AAGTTCAT" echo "$dna_seq"AAGTTCATUse parameter expansion to replace all
Ts withUto convert the DNA to RNA:echo "${dna_seq//T/U}"AAGUUCATYou can also assign the result of the parameter expansion back to a variable:
rna_seq="${dna_seq//T/U}" echo "$rna_seq"AAGUUCAT
So, the syntax for this type of parameter expansion is {var_name//<search>/replace} — let’s deconstruct that:
- Reference the variable, using the full notation, with braces (
${var_name}) - After the first two forward slashes, enter the search pattern: (
T) - After another forward slash, enter the replacement: (
U).
If you needed to replace at most one of the search patterns, a single backslash after the variable name would suffice: {var_name/<search>/replace}.
Exercise: Get the R2 file name with parameter expansion
File names of corresponding R1 and R2 FASTQ files should be identical other than the marker indicating the read direction, which is typically R1/R2 (and in some cases just 1 and 2).
Assign the file name garrigos_data/fastq/ERR10802863_R1.fastq.gz to a variable and use parameter expansion to get the name of the corresponding R2 file name. Also save the R2 file name in a variable.
Solutions
fastq_R1=garrigos_data/fastq/ERR10802863_R1.fastq.gz
fastq_R2=${fastq_R1/_R1/_R2}Above, e.g. ${fastq_R1/R1/R2}, that is without underscores, would have also worked. But note that it’s generally good to avoid unwanted search-pattern matches by making the search string as specific as possible. So perhaps ${fastq_R1/_R1.fastq.gz/_R2.fastq.gz} would have been even better.
Test that it worked:
echo "$fastq_R2"garrigos_data/fastq/ERR10802863_R2.fastq.gz5.3 A per-sample loop
We will now create a loop that:
- Loops over R1 FASTQ files only, and then infers the corresponding R2 file name.
- Defines output file names that are the same as the input file names but in a different dir.
To stay focused just on the shell syntax here, we won’t include code to run an actual bioinformatics tool (you’ll do that in this week’s exercises), but will use a fictional tool trimmer:
# [Don't run or copy this]
# Loop over the R1 files - our glob is `*_R1.fastq.gz` to only select R1 files
for R1_in in garrigos_data/fastq/*_R1.fastq.gz; do
# Get the R2 file name with parameter expansion
R2_in=${R1_in/_R1/_R2}
# Define the output files (assume that a variable $outdir exists)
R1_out="$outdir"/$(basename "$R1_in")
R2_out="$outdir"/$(basename "$R2_in")
# Report
echo "Input files: $R1_in $R2_in"
echo "Output files: $R1_out $R2_out"
# Use the imaginary program 'trimmer' with options --in1/--in2 for the R1/R2 input files,
# and --out1/--out2 for the R1/R2 output files:
trimmer --in1 "$R1_in" --in2 "$R2_in" --out1 "$R1_out" --out2 "$R2_out"
done5.4 Converting to the single-sample script format
But wait! Aren’t we supposed to write a script that only processes one sample at a time, and then run/submit that script with a loop? That’s right, so now that we know what to do, let’s switch to that setup.
Create a new script scripts/trim_mock.sh and paste the following code into it:
#!/bin/bash
set -euo pipefail
# Copy the placeholder variables
R1_in=$1
outdir=$2
# Create the output dir if needed
mkdir -p "$outdir"
# Infer the R2_in file name
R2_in=${R1_in/_R1/_R2}
# Define the output file names
R1_out="$outdir"/$(basename "$R1_in")
R2_out="$outdir"/$(basename "$R2_in")
# Initial reporting
echo "# Starting script trim_mock.sh"
date
echo "# Input R1 file: $R1_in"
echo "# Input R2 file: $R2_in"
echo "# Output R1 file: $R1_out"
echo "# Output R2 file: $R2_out"
echo
# Mock-run the tool: I preface the command with 'echo' so this will just report
# and not try to run a program that doesn't exist
echo trimmer --in1 "$R1_in" --in2 "$R2_in" --out1 "$R1_out" --out2 "$R2_out"
# Final reporting
echo
echo "# Done with script trim_mock.sh"
dateNow, add the following code to our run.sh script, and run that:
# Run the trim_mock.sh script for each sample
for R1_in in garrigos_data/fastq/*_R1.fastq.gz; do
bash scripts/trim_mock.sh "$R1_in" results/trim_mock
done# Starting script trim_mock.sh
Thu Mar 21 10:12:17 EDT 2024
# Input R1 file: garrigos_data/fastq/ERR10802863_R1.fastq.gz
# Input R2 file: garrigos_data/fastq/ERR10802863_R2.fastq.gz
# Output R1 file: results/trim_mock/ERR10802863_R1.fastq.gz
# Output R2 file: results/trim_mock/ERR10802863_R2.fastq.gz
trimmer --in1 garrigos_data/fastq/ERR10802863_R1.fastq.gz --in2 garrigos_data/fastq/ERR10802863_R2.fastq.gz --out1 results/trim_mock/ERR10802863_R1.fastq.gz --out2 results/trim_mock/ERR10802863_R2.fastq.gz
# Done with script trim_mock.sh
Thu Mar 21 10:12:17 EDT 2024
# Starting script trim_mock.sh
Thu Mar 21 10:12:17 EDT 2024
# Input R1 file: garrigos_data/fastq/ERR10802864_R1.fastq.gz
# Input R2 file: garrigos_data/fastq/ERR10802864_R2.fastq.gz
# Output R1 file: results/trim_mock/ERR10802864_R1.fastq.gz
# Output R2 file: results/trim_mock/ERR10802864_R2.fastq.gz
# [...output truncated...]Check the files in the output dir:
ls -lh results/trim_mocktotal 0
-rw-rw----+ 1 jelmer PAS0471 0 Mar 21 10:12 ERR10802863_R1.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 0 Mar 21 10:12 ERR10802863_R2.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 0 Mar 21 10:12 ERR10802864_R1.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 0 Mar 21 10:12 ERR10802864_R2.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 0 Mar 21 10:12 ERR10802865_R1.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 0 Mar 21 10:12 ERR10802865_R2.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 0 Mar 21 10:12 ERR10802866_R1.fastq.gz
# [...output truncated...]Footnotes
For a full list of installed software at OSC: https://www.osc.edu/resources/available_software/software_list.↩︎
We’ll talk more about loading (and installing) software at OSC next week.↩︎
We’ll see in the upcoming Slurm module that we when submit scripts to the OSC queue (rather than running them directly), the output of scripts that is normally printed to screen, will instead go to a sort of “log” file. So, your script’s reporting will end up in this file.↩︎
Or depending on how you use this exactly, as your notebook entry that contains the final protocol you followed.↩︎