Read QC and primer trimming
With FastQC, MultiQC, and Cutadapt
Introduction
The first series of steps our analysis workflow concerns the quality control (QC) & “pre-processing” of the raw sequence reads, which are stored in FASTQ files.
The QC part will leave the data untouched, whereas the pre-processing involves the removal of unwanted bits of sequence: in our case, amplicon primers. After the pre-processing step, we will still have FASTQ files, just with somewhat less content.
Specifically, we will go through the following steps:
- QC with FastQC
- Summarizing FastQC results with MultiQC
- Removing primers with Cutadapt
Start a new VS Code session with an open terminal:
- Log in to OSC’s OnDemand portal at https://ondemand.osc.edu.
- In the blue top bar, select
Interactive Appsand then near the bottom of the dropdown menu, clickCode Server. - In the form that appears on a new page:
- Select OSC project
PAS2714 - The starting directory:
/fs/ess/PAS2714/<user>(replace<user>with your username) Number of hours:3- Click
Launch.
- Select OSC project
- On the next page, once the top bar of the box has turned green and says
Runnning, clickConnect to VS Code. - Open a Terminal by clicking =>
Terminal=>New Terminal.
1 Running FastQC for 1 sample
1.1 Intro to FastQC
FastQC is a ubiquitous tools for quality control of FASTQ files. Running FastQC or a similar program is the first step in nearly any high-throughput sequencing project. FastQC is also a good introductory example of a tool with a command-line interface.
For each FASTQ file, FastQC outputs an HTML file that you can open in your browser with about a dozen graphs showing different QC metrics. The most important one is the per-base quality score graph shown below.
1.2 Our FASTQ files
Our FASTQ files contain reads from 2x300 bp (i.e. paired-end with 300 bp forward and 300 bp reverse reads) sequencing on an Illumina MiSeq machine.
Let’s take a look at our list of FASTQ files:
ls -lh data/fastqtotal 150M
-rw-r-----+ 1 jelmer PAS0471 2.0M Mar 1 17:09 NW102AB_R1.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 2.6M Mar 1 17:09 NW102AB_R2.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 2.3M Mar 1 17:09 NW102C_R1.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 3.0M Mar 1 17:09 NW102C_R2.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 1.9M Mar 1 17:09 NW103AB_R1.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 2.6M Mar 1 17:09 NW103AB_R2.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 2.3M Mar 1 17:09 NW103C_R1.fastq.gz
-rw-r-----+ 1 jelmer PAS0471 3.1M Mar 1 17:09 NW103C_R2.fastq.gz
# [...output truncated...]Note in the file listing above that:
- There are two files per sample:
_R1(forward reads) and_R2(reverse reads). This indicates that we have data from paired-end reads, as is customary when doing amplicon metabarcoding. - The files all have a
.gzextension, indicating they have been compressed with the gzip utility.
Run the following two cp commands to copy the necessary data:
cp -rv /fs/ess/PAS2714/share/data /fs/ess/PAS2714/users/$USER
cp -rv /fs/ess/PAS2714/share/results /fs/ess/PAS2714/users/$USER1.3 Building our FastQC command
To run FastQC, we can use the command fastqc.
If you want to analyze one of your FASTQ files with default FastQC settings, a complete FastQC command to do so would simply be fastqc followed by the name of the file:
# (Don't run this)
fastqc data/fastq/NW102AB_R1.fastq.gzHowever, an annoying FastQC default behavior is that it writes its output files in the dir where the input files are — in general, it’s not great practice to directly mix your primary data and results like that!
To figure out how we can change that behavior, first consider that many commands and bioinformatics tools alike have an option -h and/or --help to print usage information to the screen. Let’s try that:
fastqc -hbash: fastqc: command not found...However, there is a wrinkle: while FastQC is installed at OSC1, we have to first “load it”. The way we will do this here is with a a so-called “Conda environment” that has FastQC installed along with the other programs we will need today.
Here’s how we can load that Conda software environment — we first load OSC’s (mini)conda installation, and then we can load (“activate”) the Conda environment that I created for you:
module load miniconda3
source activate /fs/ess/PAS0471/jelmer/conda/mbar24We won’t have time to get into this now, but you want to learn more about Conda / software usage at supercomputers, see this reference page elsewhere on the website.
Exercise: FastQC help and output dir
Print FastQC’s help info, and figure out which option you can use to specify a custom output directory.
Click for the solution
fastqc -h and fastqc --help will both work to show the help info.
You’ll get quite a bit of output printed to screen, including the snippet about output directories that is reproduced below:
fastqc -h -o --outdir Create all output files in the specified output directory.
Please note that this directory must exist as the program
will not create it. If this option is not set then the
output file for each sequence file is created in the same
directory as the sequence file which was processed.So, you can use -o or equivalently, --outdir to specify an output dir.
With the added --outdir (or -o) option, let’s try to run the following FastQC command:
# We'll have to first create the outdir ourselves, in this case
mkdir -p results/fastqc
# Now we run FastQC
fastqc --outdir results/fastqc data/fastq/NW102AB_R1.fastq.gzapplication/gzip
Started analysis of NW102AB_R1.fastq.gz
Approx 5% complete for NW102AB_R1.fastq.gz
Approx 10% complete for NW102AB_R1.fastq.gz
Approx 15% complete for NW102AB_R1.fastq.gz
[...truncated...]
Analysis complete for NW102AB_R1.fastq.gzSuccess!! 🎉
1.4 FastQC output files
Let’s take a look at the files in the output dir we specified:
ls -lh results/fastqctotal 1.2M
-rw-r--r-- 1 jelmer PAS0471 241K Mar 13 14:50 NW102AB_R1_fastqc.html
-rw-r--r-- 1 jelmer PAS0471 256K Mar 13 14:50 NW102AB_R1_fastqc.zip- There is a
.zipfile, which contains tables with FastQC’s data summaries. - There is an
.html(HTML) file, which contains plots — this is what we’ll look at next.
Exercise: Another FastQC run
Run FastQC for the corresponding R2 FASTQ file. Would you use the same output dir?
Click for the solution
Yes, it makes sense to use the same output dir, since as you could see above, the output file names have the input file identifiers in them. As such, we don’t need to worry about overwriting files, and it will be more convenient to have all results in a single dir.
To run FastQC for the R2 (reverse-read) file:
fastqc --outdir results/fastqc data/fastq/NW102AB_R2.fastq.gzStarted analysis of NW102AB_R2.fastq.gz
Approx 5% complete for NW102AB_R2.fastq.gz
Approx 10% complete for NW102AB_R2.fastq.gz
Approx 15% complete for NW102AB_R2.fastq.gz
[...truncated...]
Analysis complete for NW102AB_R2.fastq.gzls -lh results/fastqc-rw-r--r-- 1 jelmer PAS0471 241K Mar 13 14:50 NW102AB_R1_fastqc.html
-rw-r--r-- 1 jelmer PAS0471 256K Mar 13 14:50 NW102AB_R1_fastqc.zip
-rw-r--r-- 1 jelmer PAS0471 234K Mar 13 14:53 NW102AB_R2_fastqc.html
-rw-r--r-- 1 jelmer PAS0471 244K Mar 13 14:53 NW102AB_R2_fastqc.zipNow, we have four files: two for each of our preceding successful FastQC runs.
2 Interpreting FastQC output
2.1 FastQC HTML modules
We’ll now go through a couple of the FastQC plots/modules with example plots2 with good/bad results for reference.
FastQC has “pass” (checkmark in green), “warning” (exclamation mark in orange), and “fail” (cross in red) assessments for each module. These assessments are handy, but a “warning”/“fail” is not necessarily the bad news it may appear to be:
- Some of these modules are perhaps overly strict.
- Some warnings and fails are easily remedied or simply not a very big deal.
- FastQC effectively assumes that your data is derived from whole-genome shotgun sequencing — some other types of data with different properties will therefore always trigger a couple of warnings and fails, but these are not meaningful. This is very much the case for metabarcoding data.

The green checkmarks indicate Pass, the orange exclamation marks indicate Warning, and the red crosses indicate Fail.
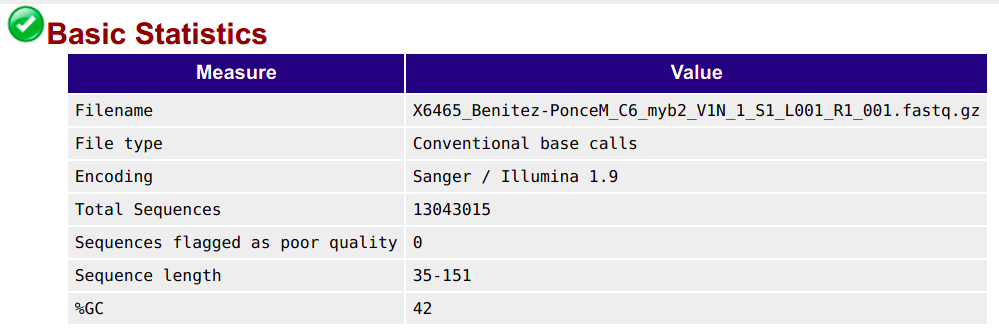
Basic statistics
This contains, among other things, the number of sequences (reads) and the read length range:
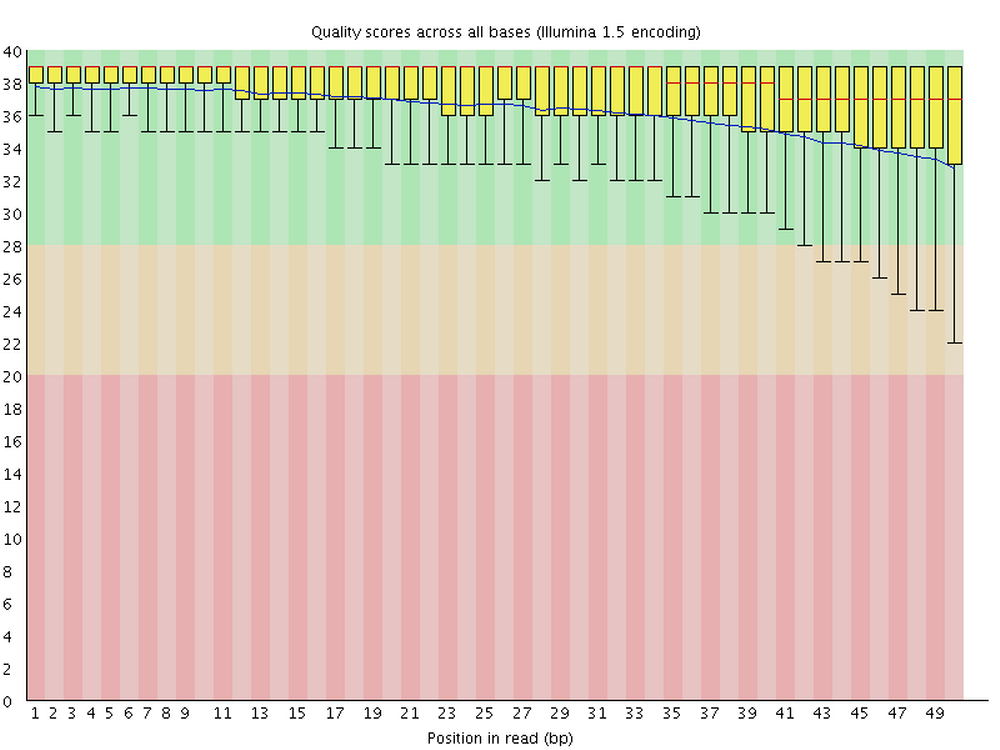
Per base quality sequence quality
This figure visualizes the mean per-base quality score (y-axis) along the length of the reads (x-axis). Note that:
- A decrease in sequence quality along the reads (from left to right) is normal.
- R2 (reverse) reads are usually of worse quality than R1 (forward) reads.
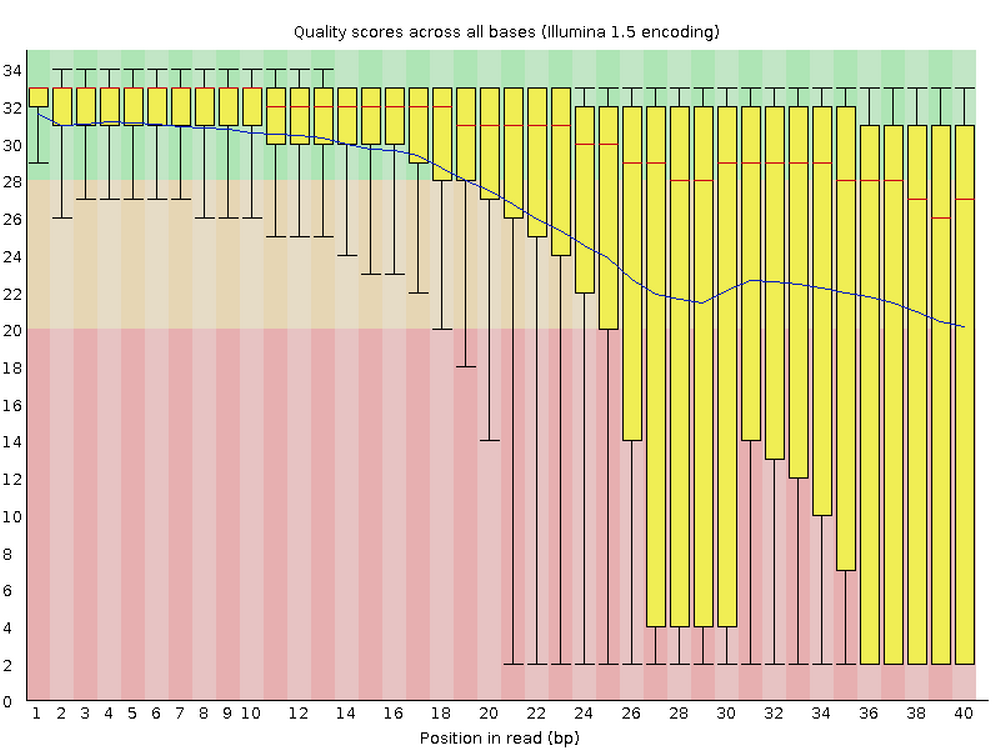
To interpret the y-axis quality scores, note the color scaling in the graphs above, and see this table for details:
| Phred quality score | Error probability | Rough interpretation |
|---|---|---|
| 10 | 1 in 10 | terrible |
| 20 | 1 in 100 | bad |
| 30 | 1 in 1,000 | good |
| 40 | 1 in 10,000 | excellent |
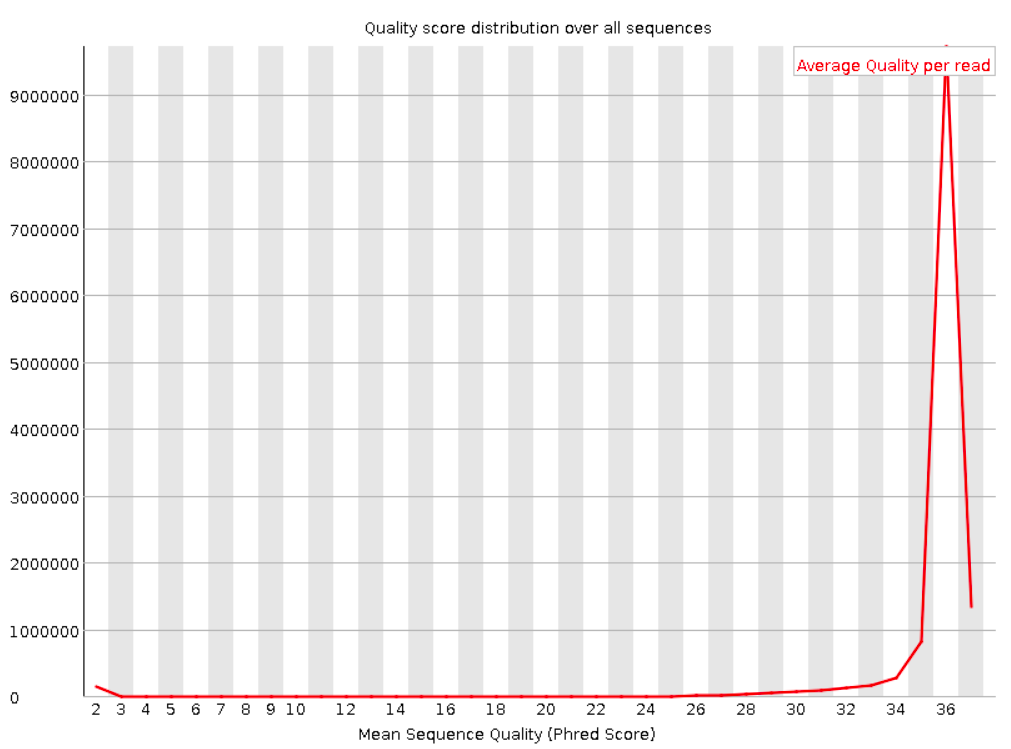
Per sequence quality scores
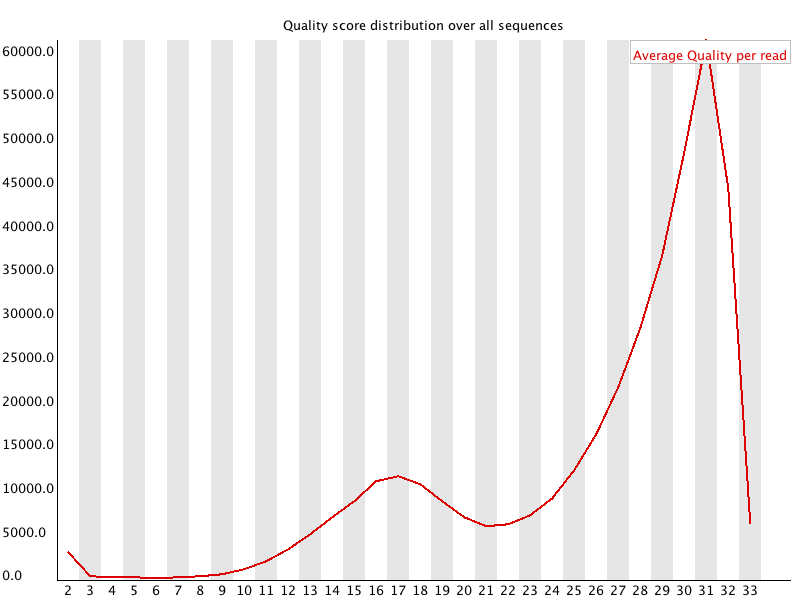
This shows the same quality scores we saw above, but now simply as a density plot of per-read averages, with the quality score now along the x-axis, and the number of reads with that quality score along the y-axis:
Per tile sequence quality
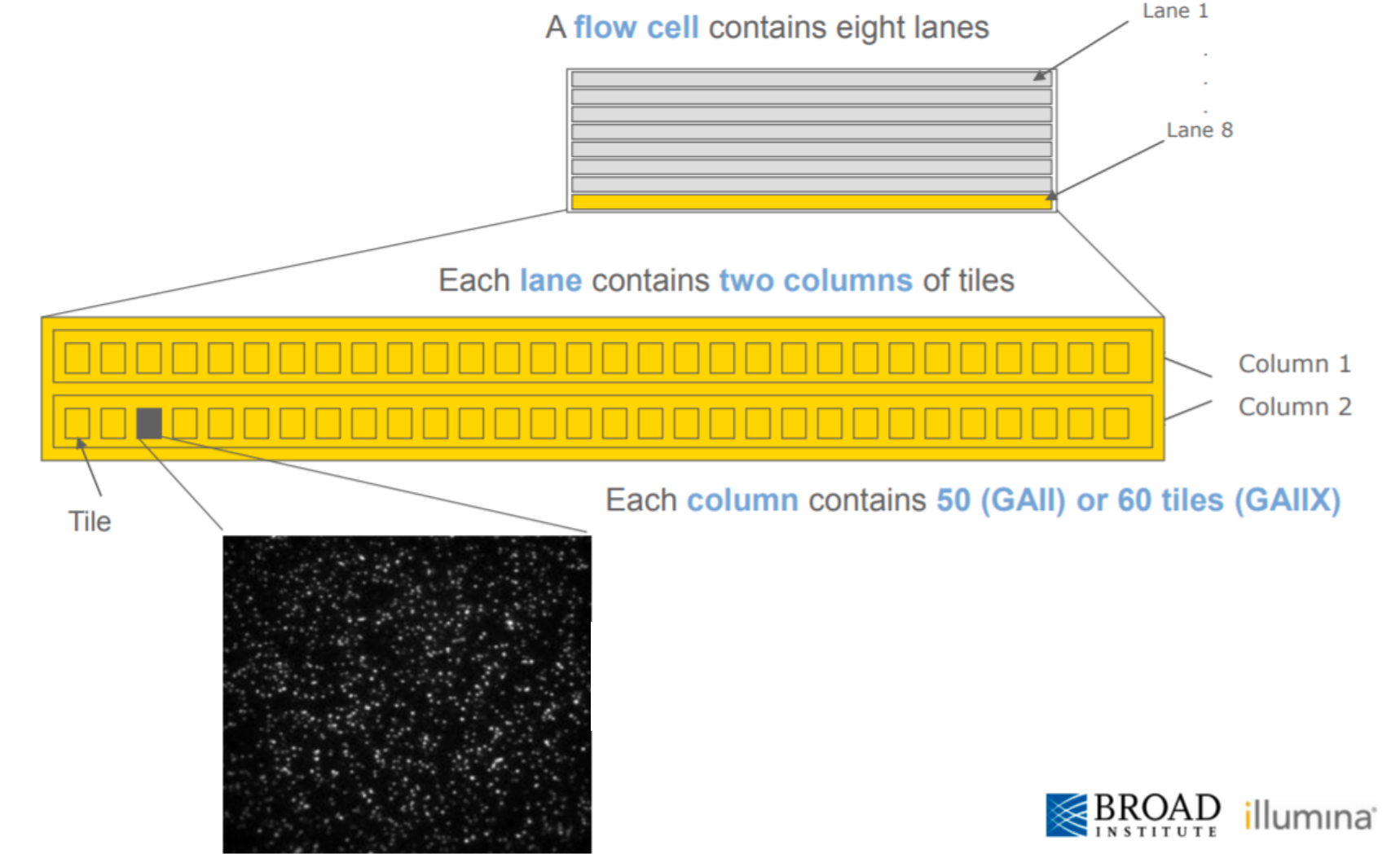
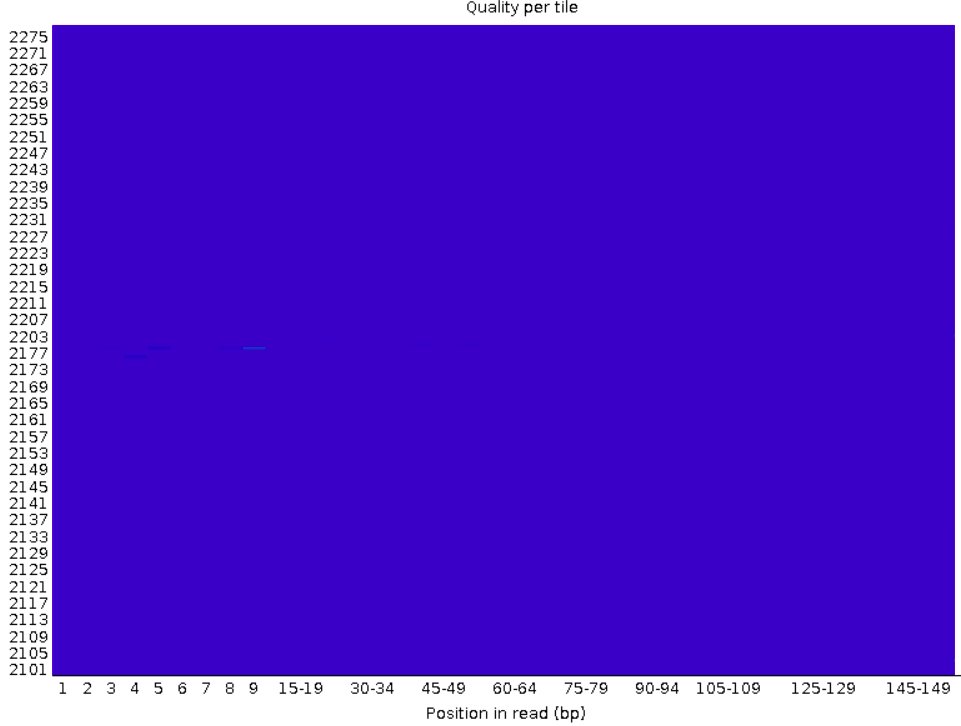
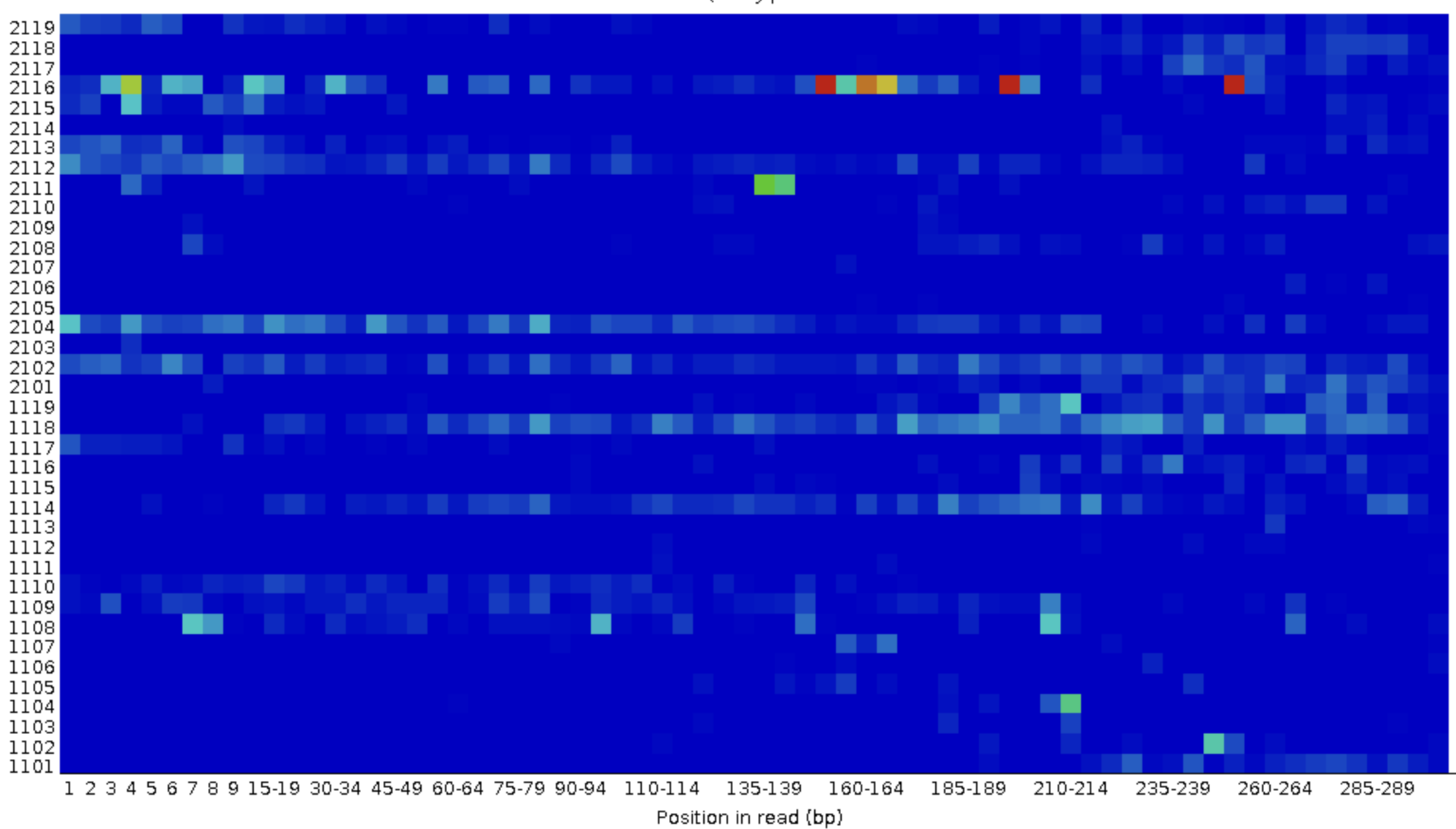
This graph shows whether specific tiles (areas on the flow cell, see box below) have lower quality bases in some parts of or across all of the read.
Poor qualities in some but not other tiles may indicate some (transient) problems with the flow cell. For example, if a tile has poor qualities for a stretch of bases, this could indicate that there was a bubble. If such tile-based problems are severe, you can contact the sequencing facility as they may be able to re-run your data at no cost.
Sequence length distribution
With Illumina sequencing, the vast majority of reads typically have nearly the same read length — in our case, 300 bp. This graph can help you check if that is indeed the case. The module will throw a warning as soon as not all sequences are of the same length (like below), but having reads with a slightly shorter read length is normal and does not matter.
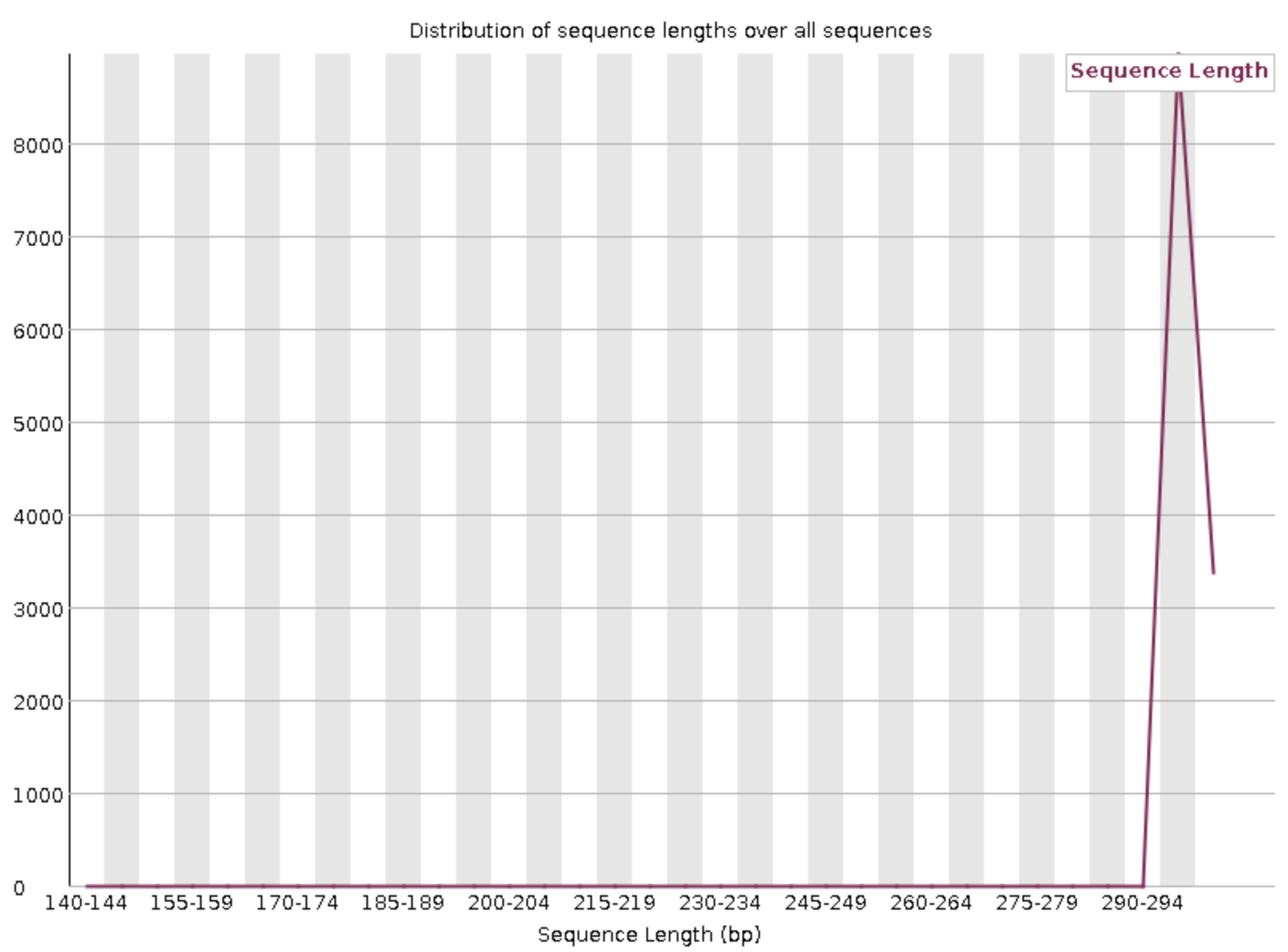
Nearly all reads have the same read length.
FastQC still threw a warning.
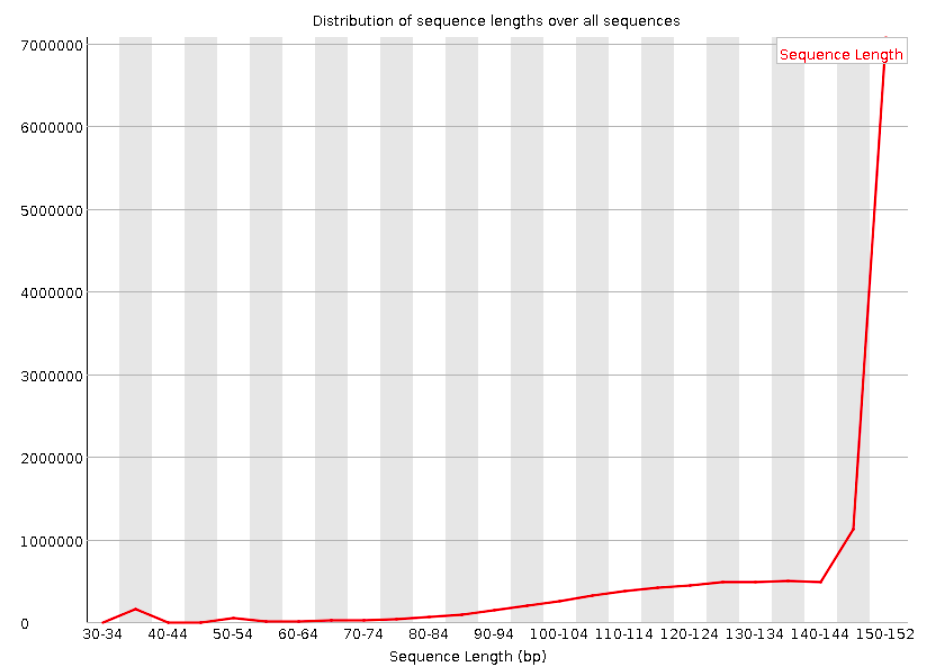
There are unusually many reads that are shorter than the aimed-for, 150 bp read length. Though the large majority of reads are still 150 bp.
Other FastQC modules
Another module that can help to check the overall quality of your reads is Per base N content: its line graph should typically not visibly rise above 0%.
The remaining modules are not that useful for metabarcoding data:
2.2 Checking your FastQC results
First, you’ll unfortunately have to download FastQC’s output HTML files to your computer to be able to view them:
- Find the FastQC HTML files in the file explorer in the VS Code side bar.
- Right-click on one of them, click
Download...and follow the prompt to download the file somewhere to your computer (doesn’t matter where). - Repeat this for the second file.
- Then, open your computer’s file browser, find the downloaded files, and double-click on one. It should be opened in your default web browser.
Alternatively, you can take a look at the FastQC output files on this website: R1 and R2.
Exercise: Interpreting your FastQC results
Open the HTML file for the R1 FASTQ file and go through the modules we discussed above. Can you make sense of it? Does the data look good to you, overall?
Now open the HTML file for the R2 FASTQ file and take a look just at the quality scores. Does it look any worse than the R1?
3 Running FastQC for all samples
If we want to run FastQC for all samples, it will be much better to write a shell script and submit that as a so-called Slurm batch job, rather than running FastQC “interactively” like we did for the first sample.
This is especially true for a complete data set, which would have much larger FASTQ files and possibly more samples.
3.1 The components of our script
Here is all the code we needed for out first FastQC run:
# Load the software
module load miniconda3
source activate /fs/ess/PAS0471/jelmer/conda/mbar24
# Create the output dir
mkdir -p results/fastqc
# Run FastQC
fastqc --outdir results/fastqc data/fastq/NW102AB_R1.fastq.gzWe will need that same code in the script, except that we will need to modify our call to fastqc — we will loop over all FASTQ files as follows:
# Run FastQC (replacement for fastqc line above)
for fastq_file in data/fastq/*fastq.gz; do
fastqc --outdir results/fastqc "$fastq_file"
done- We are looping over all FASTQ files with the globbing pattern
data/fastq/*fastq.gz. The loop will run as many times as we have FASTQ files. - In every iteration of the loop, the
"$fastq_file"variable will contain 1 FASTQ file name, and we will runfastqcfor that file7.
We will be submitting this script as a batch job to the Slurm compute job scheduler. To do so, we should also add some lines at the top of the script:
#!/bin/bash
#SBATCH --account=PAS2714
#SBATCH --output=slurm-fastqc.out- The first line
#!/bin/bashmerely indicates that this is a shell script8 rather than, say, an R or Python script. - The lines starting with
#SBATCHtell Slurm some details about our compute job request (much like we did when we filled out the form to start a VS Code session):- We always need to specify an “
account”, i.e. OSC project, that should be billed. - The only other option (of many possible!) we will use here is to specify the
outputfile: this is where any output will go that would otherwise be printed to screen, such as the FastQC progress output we saw above.
- We always need to specify an “
We will also add the following line to change some shell script settings, which will cause the script to stop running if any errors occur:
# Strict bash settings
set -euo pipefail3.2 Our final script
- Open a new file in VS Code: click , then
File, thenNew File. - Save the file (e.g. press Ctrl/⌘+S) in your
scriptsdirectory asfastqc.sh. - Paste the following code in the script:
#!/bin/bash
#SBATCH --account=PAS2714
#SBATCH --output=slurm-fastqc.out
# Strict bash settings
set -euo pipefail
# Load the software
module load miniconda3
source activate /fs/ess/PAS0471/jelmer/conda/mbar24
# Create the output dir
mkdir -p results/fastqc
# Run FastQC for all FASTQ files
for fastq_file in data/fastq/*fastq.gz; do
fastqc --outdir results/fastqc "$fastq_file"
done
# Report
echo "Done with script fastqc.sh"
date3.3 Submitting the script
Submit the script to Slurm (“submit it to the queue”) with the sbatch command:
sbatch scripts/fastqc.shSubmitted batch job 27047185After some seconds (sometimes up to a minute or so9), the Slurm job should start and create the output file that we specified at the top of the script: slurm-fastqc.out.
3.4 Checking the output
In our earlier two FastQC runs, FastQC logging output (“10% complete”, etc.) was printed to screen. But because this job now runs remotely on another compute node, such output will end up in the “Slurm log file” whose name we specified in the script. Let’s take a look:
less slurm-fastqc.outapplication/gzip
Started analysis of NW102AB_R1.fastq.gz
Approx 5% complete for NW102AB_R1.fastq.gz
Approx 15% complete for NW102AB_R1.fastq.gz
Approx 20% complete for NW102AB_R1.fastq.gz
Approx 30% complete for NW102AB_R1.fastq.gz
Approx 35% complete for NW102AB_R1.fastq.gz
Approx 45% complete for NW102AB_R1.fastq.gz
Approx 50% complete for NW102AB_R1.fastq.gz
Approx 60% complete for NW102AB_R1.fastq.gz
Approx 70% complete for NW102AB_R1.fastq.gz
Approx 75% complete for NW102AB_R1.fastq.gz
Approx 85% complete for NW102AB_R1.fastq.gz
Approx 90% complete for NW102AB_R1.fastq.gz
Analysis complete for NW102AB_R1.fastq.gz
application/gzip
Started analysis of NW102AB_R2.fastq.gz
Approx 5% complete for NW102AB_R2.fastq.gz
Approx 15% complete for NW102AB_R2.fastq.gz
Approx 20% complete for NW102AB_R2.fastq.gz
#[...output truncated...]That looks good, in the output I printed above we can see that FastQC ran to completion for one FASTQ file and then started a second — and this will go on and on, for all of our 64 FASTQ files.
You will know that the job has successfully finished when the last few lines of the Slurm log file read “Done with script fastqc.sh” and print the date and time (as per the last lines of our script!):
tail slurm-fastqc.outApprox 75% complete for W404BC_R2.fastq.gz
Approx 85% complete for W404BC_R2.fastq.gz
Approx 90% complete for W404BC_R2.fastq.gz
Approx 95% complete for W404BC_R2.fastq.gz
Analysis complete for W404BC_R2.fastq.gz
Done with script fastqc.sh
Wed Mar 6 13:34:10 EST 2024Of course, we should also check the main output files — the HTMLs and zip files:
ls results/fastqcNW102AB_R1_fastqc.html NW103C_R1_fastqc.zip NW203A_R2_fastqc.html NW304BC_R2_fastqc.zip NW403BC_R1_fastqc.html W101AB_R1_fastqc.zip W103C_R2_fastqc.html W205A_R2_fastqc.zip W304AB_R1_fastqc.html W403C_R1_fastqc.zip
NW102AB_R1_fastqc.zip NW103C_R2_fastqc.html NW203A_R2_fastqc.zip NW305AB_R1_fastqc.html NW403BC_R1_fastqc.zip W101AB_R2_fastqc.html W103C_R2_fastqc.zip W205BC_R1_fastqc.html W304AB_R1_fastqc.zip W403C_R2_fastqc.html
NW102AB_R2_fastqc.html NW103C_R2_fastqc.zip NW203BC_R1_fastqc.html NW305AB_R1_fastqc.zip NW403BC_R2_fastqc.html W101AB_R2_fastqc.zip W204A_R1_fastqc.html W205BC_R1_fastqc.zip W304AB_R2_fastqc.html W403C_R2_fastqc.zip
NW102AB_R2_fastqc.zip NW201AB_R1_fastqc.html NW203BC_R1_fastqc.zip NW305AB_R2_fastqc.html NW403BC_R2_fastqc.zip W101C_R1_fastqc.html W204A_R1_fastqc.zip W205BC_R2_fastqc.html W304AB_R2_fastqc.zip W404A_R1_fastqc.html
NW102C_R1_fastqc.html NW201AB_R1_fastqc.zip NW203BC_R2_fastqc.html NW305AB_R2_fastqc.zip NW404A_R1_fastqc.html W101C_R1_fastqc.zip W204A_R2_fastqc.html W205BC_R2_fastqc.zip W304C_R1_fastqc.html W404A_R1_fastqc.zip
NW102C_R1_fastqc.zip NW201AB_R2_fastqc.html NW203BC_R2_fastqc.zip NW305C_R1_fastqc.html NW404A_R1_fastqc.zip W101C_R2_fastqc.html W204A_R2_fastqc.zip W303AB_R1_fastqc.html W304C_R1_fastqc.zip W404A_R2_fastqc.html
NW102C_R2_fastqc.html NW201AB_R2_fastqc.zip NW304A_R1_fastqc.html NW305C_R1_fastqc.zip NW404A_R2_fastqc.html W101C_R2_fastqc.zip W204BC_R1_fastqc.html W303AB_R1_fastqc.zip W304C_R2_fastqc.html W404A_R2_fastqc.zip
NW102C_R2_fastqc.zip NW201C_R1_fastqc.html NW304A_R1_fastqc.zip NW305C_R2_fastqc.html NW404A_R2_fastqc.zip W103AB_R1_fastqc.html W204BC_R1_fastqc.zip W303AB_R2_fastqc.html W304C_R2_fastqc.zip W404BC_R1_fastqc.html
NW103AB_R1_fastqc.html NW201C_R1_fastqc.zip NW304A_R2_fastqc.html NW305C_R2_fastqc.zip NW404BC_R1_fastqc.html W103AB_R1_fastqc.zip W204BC_R2_fastqc.html W303AB_R2_fastqc.zip W403AB_R1_fastqc.html W404BC_R1_fastqc.zip
NW103AB_R1_fastqc.zip NW201C_R2_fastqc.html NW304A_R2_fastqc.zip NW403A_R1_fastqc.html NW404BC_R1_fastqc.zip W103AB_R2_fastqc.html W204BC_R2_fastqc.zip W303C_R1_fastqc.html W403AB_R1_fastqc.zip W404BC_R2_fastqc.html
NW103AB_R2_fastqc.html NW201C_R2_fastqc.zip NW304BC_R1_fastqc.html NW403A_R1_fastqc.zip NW404BC_R2_fastqc.html W103AB_R2_fastqc.zip W205A_R1_fastqc.html W303C_R1_fastqc.zip W403AB_R2_fastqc.html W404BC_R2_fastqc.zip
NW103AB_R2_fastqc.zip NW203A_R1_fastqc.html NW304BC_R1_fastqc.zip NW403A_R2_fastqc.html NW404BC_R2_fastqc.zip W103C_R1_fastqc.html W205A_R1_fastqc.zip W303C_R2_fastqc.html W403AB_R2_fastqc.zip
NW103C_R1_fastqc.html NW203A_R1_fastqc.zip NW304BC_R2_fastqc.html NW403A_R2_fastqc.zip W101AB_R1_fastqc.html W103C_R1_fastqc.zip W205A_R2_fastqc.html W303C_R2_fastqc.zip W403C_R1_fastqc.htmlThat’s a lot of files! Do we need to check all of them? Luckily not, thanks to MultiQC.
4 Summarizing QC results with MultiQC
Here are some challenges you may run into after running FastQC:
- When you have many FASTQ files, you’ll generate a lot of FastQC HTML files to sort through (as we did above).
- Even if you diligently go through each file, it’s not that easy to compare the results across samples in detail, since they are not drawn in the same graphs.
MultiQC addresses these problems by aggregating FastQC results from many files, and summarizing them into a single HTML file with (still) one graph per FastQC module.
MultiQC’s graphs are also interactive, but here is a static example of a graph showing the mean base quality scores along the read for many FASTQ files:
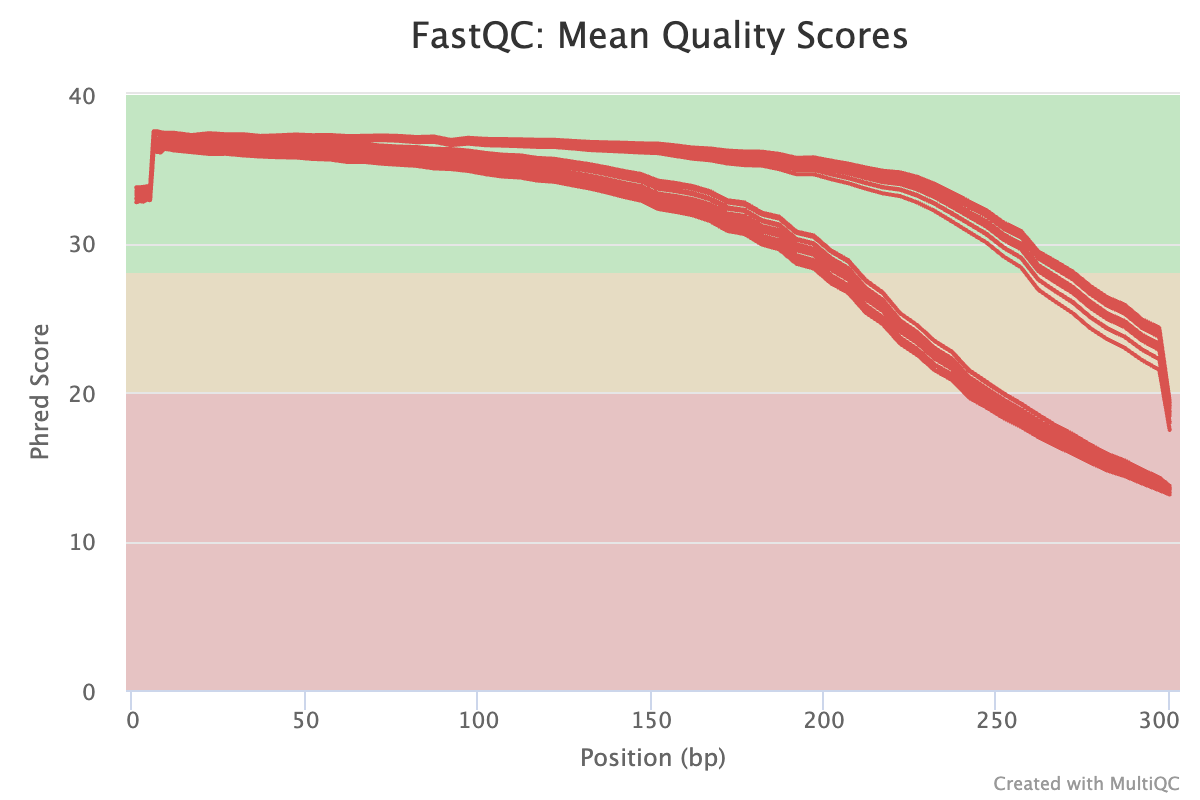
Above, what could the two “groups of lines”, which diverge towards the right-hand side, represent?
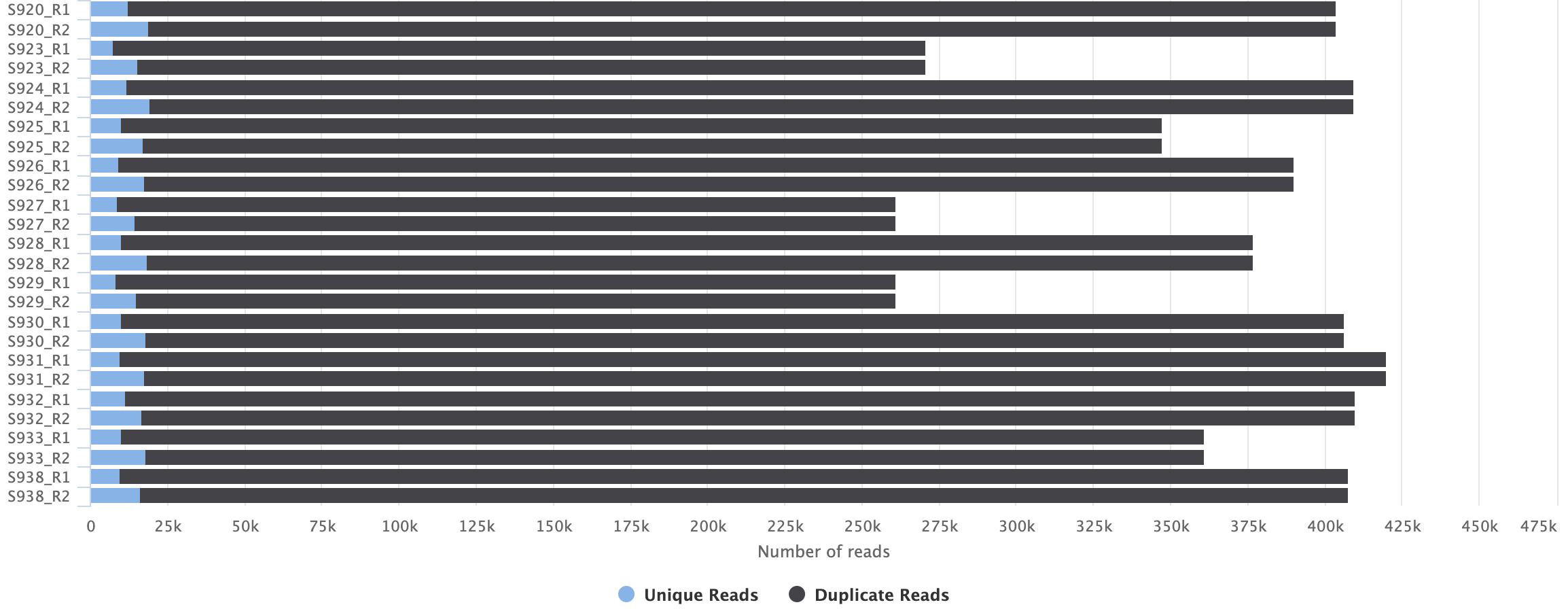
These are the files with forward (top lines, better quality) and reverse (bottom lines, worse quality) reads.MultiQC will also create a graph comparing the number of reads across files, which can be quite useful:
4.1 Running MultiQC
We will only need to run MultiQC once (because it will aggregate all FastQC results at once), and that will only take a few seconds — therefore, we can run the command interactively without using a script.
We can check MultiQC’s help with the --help option:
multiqc --help
# (Only the top part of the output is shown in the screenshot below)
As the first couple of help lines in the paler gray color explain, MultiQC will search the [ANALYSIS DIRECTORY], a dir that we pass to it as an argument at the end of the command line.
If we tell MultiQC (command multiqc) about the results/fastqc directory like so, it should find and then aggregate all the FastQC results in there:
# (Don't run this - we'll complete the command in a second)
multiqc results/fastqcIf you don’t/no longer have the mbar24 Conda environment active, (re-)activate it as follows:
module load miniconda3
source activate /fs/ess/PAS0471/jelmer/conda/mbar24The default output directory of MultiQC is the current working directory, so just like with FastQC, we do want to use the option for the output dir — this is our final command and you can go ahead and execute it:
# Run MultiQC to summarize the FastQC results
multiqc --outdir results/multiqc results/fastqc
4.2 MultiQC output
Once its done, you should have the following files in the output dir:
ls -lh results/multiqctotal 1.7M
drwxr-xr-x 2 jelmer PAS2250 4.0K Mar 13 14:57 multiqc_data
-rw-r--r-- 1 jelmer PAS2250 1.7M Mar 13 14:57 multiqc_report.htmlGo ahead and find the multiqc_report.html listed above in VS Code’s file browser, right-click on it and download it to your computer. Then, click on the file in your own computer to open it in your browser (i.e., just like we did with the FastQC output).
You can also find a copy of the MultiQC HTML output file here.
Exercise: Explore the MultiQC results
Check for example whether patterns are consistent across samples, or if there are any outliers.
5 Cutadapt
When you prepare samples for amplicon metabarcoding, you amplify a specific region with primers, and these primers will be included in the sequences that you receive. Before we go any further, we need to remove these primer sequences, which we can do with the program Cutadapt.
We will write a script with a loop to run Cutadapt for all samples and submit it as a batch job like we did with FastQC.
5.1 Primer sequences
When we run Cutadapt, we need to tell it about our primer sequences as well as their reverse complements. We’ll start by storing our particular primer sequences in variables:
# Primer sequences
primer_f=GTGTGYCAGCMGCCGCGGTAA
primer_r=GGACTACNVGGGTWTCTAATNote that the Y, M, N, V and W in the primer sequences are so-called “ambiguity codes” that represent multiple possible bases. For example, a Y represents a C or a T, and an N represents any of the 4 bases.
There are many ways of getting the reverse complement of a sequence, including manually building it up, but here we’ll use a trick with the tr command to change each base into its complement, followed by the rev command to get the reverse complement — for example, for the forward primer:
echo "$primer_f" | tr ATCGYRKMBDHV TAGCRYMKVHDB | revTTACCGCGGCKGCTGRCACACBelow, we’ll get the reverse complement for both primers, and will store those in a variable as well using the construct variable=$(command)10:
# Get the reverse-complements of the primers
primer_f_rc=$(echo "$primer_f" | tr ATCGYRKMBDHV TAGCRYMKVHDB | rev)
primer_r_rc=$(echo "$primer_r" | tr ATCGYRKMBDHV TAGCRYMKVHDB | rev)
# Check the sequences
echo "$primer_f_rc"
echo "$primer_r_rc"TTACCGCGGCKGCTGRCACAC
ATTAGAWACCCBNGTAGTCCtr | rev command (Click to expand)
tr A Tchanges every A to a Ttr ATC TAGchanges every A to a T (first character in each of the two sequences), every T to an a (second character in each of the two sequences), and every C to a G (third character in each of the two sequences).- Therefore, in the full
trcommand above, we list all possible bases and ambiguity codes, and then change them to their complement. - The
revcommand simply reverses a sequence, so that we end up with the reverse complement.
echo ACCT | revTCCA5.2 Building the Cutadapt command
First, here is how we can tell Cutadapt about the primer sequences:
- With the
-aand-Aoption we specify the primer sequences in the forward and reverse reads, respectively. - The forward reads should contain the forward primer at the beginning of the read. Because reads are sometimes longer than the amplicon length, the reverse primer may be present at the end of the read, but as its reverse complement. We specify this using
-a "$primer_f"..."$primer_r_rc". - Similarly, the reverse reads should contain the reverse primer at the beginning of the read, and may contain the reverse complement of the forward primer at the end of the read, which we specify using
-A "$primer_r"..."$primer_f_rc".
All in all, our primer specification looks like this:
# (Don't run this - this will be part of our script)
cutadapt \
-a "$primer_f"..."$primer_r_rc" \
-A "$primer_r"..."$primer_f_rc"\
Above, I spread the command across multiple lines, which makes it a little easier to read. You can run the command exactly like that: the backslashes (\) at the end of all except the last line tell the shell that our command will continue on the next line.
We will also:
- Tell Cutadapt to only keep sequences that contain the primer11, with the
--trimmed-onlyoption. - Instruct Cutadapt to use 8 “cores” with
--cores 8, which can speed up the run by up to 8-fold. For our small FASTQ files, this isn’t really necessary, but for a larger dataset, that can save quite some time.
# (Don't run this - this will be part of our script)
cutadapt \
-a "$primer_f"..."$primer_r_rc" \
-A "$primer_r"..."$primer_f_rc" \
--trimmed-only \
--cores 8Finally, let’s also add the output files (--output for R1 and --paired-output for R2) and the input files (as positional arguments at the end of the command) for a single example sample. With that, we have a final example command of running Cutadapt for a single sample:
# (Don't run this - this will be part of our script)
cutadapt \
-a "$primer_f"..."$primer_r_rc" \
-A "$primer_r"..."$primer_f_rc" \
--trimmed-only \
--cores 8 \
--output results/cutadapt/NW102AB_R1.fastq.gz \
--paired-output results/cutadapt/NW102AB_R1.fastq.gz \
data/fastq/NW102AB_R1.fastq.gz \
data/fastq/NW102AB_R2.fastq.gz5.3 Our Cutadapt loop
In our script, we will run Cutadapt inside a loop, similar to how we ran FastQC. However, this case is a bit more complicated, because we need to run Cutadapt for one sample and therefore two FASTQ files at a time, rather than for one FASTQ file at a time.
We will do that by looping over the R1 (forward read) files only, and inside the loop, inferring the name of the R2 file:
# (Don't run this - this will be part of our script)
# Loop over the R1 files
for R1_in in data/fastq/*R1.fastq.gz; do
# Get the R2 file name with "parameter expansion"
# This does a search-and-replace: replace "_R1" with "_R2"
R2_in=${R1_in/_R1/_R2}
# Report
echo "Input files: $R1_in $R2_in"
# Define the output files
R1_out="$outdir"/$(basename "$R1_in")
R2_out="$outdir"/$(basename "$R2_in")
# Run Cutadapt
cutadapt \
-a "$primer_f"..."$primer_r_rc" \
-A "$primer_r"..."$primer_f_rc" \
--trimmed-only \
--cores 8 \
--output "$R1_out" \
--paired-output "$R2_out" \
"$R1_in" "$R2_in"
done5.4 The final script
Open a new text file and save it as scripts/cutadapt.sh. Then paste the following code into the script:
#!/bin/bash
#SBATCH --account=PAS2714
#SBATCH --output=slurm-cutadapt.out
#SBATCH --cpus-per-task=8
# Strict bash settings
set -euo pipefail
# Load the software
module load miniconda3
source activate /fs/ess/PAS0471/jelmer/conda/mbar24
# Primer sequences
primer_f=GTGTGYCAGCMGCCGCGGTAA
primer_r=GGACTACNVGGGTWTCTAAT
# Get the reverse-complements of the primers
primer_f_rc=$(echo "$primer_f" | tr ATCGYRKMBDHV TAGCRYMKVHDB | rev)
primer_r_rc=$(echo "$primer_r" | tr ATCGYRKMBDHV TAGCRYMKVHDB | rev)
# Create the output dir
outdir=results/cutadapt
mkdir -p "$outdir"
# Loop over the R1 files
for R1_in in data/fastq/*R1.fastq.gz; do
# Get the R2 file name
R2_in=${R1_in/_R1/_R2}
# Report
echo "Input files: $R1_in $R2_in"
# Define the output files
R1_out="$outdir"/$(basename "$R1_in")
R2_out="$outdir"/$(basename "$R2_in")
# Run Cutadapt
cutadapt \
-a "$primer_f"..."$primer_r_rc" \
-A "$primer_r"..."$primer_f_rc" \
--trimmed-only \
--cores 8 \
--output "$R1_out" \
--paired-output "$R2_out" \
"$R1_in" "$R2_in"
done
# Report
echo "Done with script cutadapt.sh"
dateNow we submit the script as a batch job in the same way we did with the FastQC script:
sbatch scripts/cutadapt.shSubmitted batch job 270472475.5 Check the output
Once we see the “Done with script” line when we use tail on the Slurm log file, we know the job has finished:
tail slurm-cutadapt.out22 4 0.0 2 0 3 1
24 1 0.0 2 0 1
25 1 0.0 2 0 0 1
34 1 0.0 2 0 0 1
38 2 0.0 2 0 2
42 1 0.0 2 0 0 1
44 1 0.0 2 0 1
47 1 0.0 2 0 1
Done with script cutadapt.sh
Wed Mar 6 14:46:56 EST 2024Let’s use less to take a closer look at the logging output:
less slurm-cutadapt.outInput files: data/fastq/NW102AB_R1.fastq.gz data/fastq/NW102AB_R2.fastq.gz
This is cutadapt 4.6 with Python 3.10.13
Command line parameters: -a GTGTGYCAGCMGCCGCGGTAA...ATTAGAWACCCBNGTAGTCC -A GGACTACNVGGGTWTCTAAT...TTACCGCGGCKGCTGRCACAC --trimmed-only --cores 8 --output results/cutadapt/NW102AB_R1.fastq.gz --paired-output results/cutadapt/NW102AB_R2.fast
q.gz data/fastq/NW102AB_R1.fastq.gz data/fastq/NW102AB_R2.fastq.gz
Processing paired-end reads on 8 cores ...
Finished in 0.875 s (68.148 µs/read; 0.88 M reads/minute).
=== Summary ===
Total read pairs processed: 12,844
Read 1 with adapter: 12,798 (99.6%)
Read 2 with adapter: 12,541 (97.6%)
== Read fate breakdown ==
Pairs discarded as untrimmed: 337 (2.6%)
Pairs written (passing filters): 12,507 (97.4%)
# [...output truncated...]We can see that Cutadapt reports the numbers and percentages of reads that contained what it calls the “adapter”: in our case, that’s the primer. In the example above, and that is typical, the percentages are in the upper 90s.
We will use the grep command to print all lines that contain the information on numbers and percentages of reads with the primer sequences:
grep "with adapter:" slurm-cutadapt.out Read 1 with adapter: 12,798 (99.6%)
Read 2 with adapter: 12,541 (97.6%)
Read 1 with adapter: 14,499 (99.7%)
Read 2 with adapter: 14,211 (97.7%)
Read 1 with adapter: 12,174 (99.7%)
Read 2 with adapter: 11,835 (97.0%)
Read 1 with adapter: 15,054 (99.7%)
Read 2 with adapter: 14,737 (97.6%)
# [...output truncated...]You should always take a careful look at this output, to check if there are no samples with much lower percentages: it looks like there are no such samples in this case, fortunately.
Use this command to extract and sort the percentages:
grep "with adapter:" slurm-cutadapt.out | cut -d"(" -f2 | sort -n | head97.0%)
97.3%)
97.5%)
97.5%)
97.5%)
97.5%)
97.6%)Use this command to print the 7 lines preceding each match, so you can see the file names (samples) that correspond to each percentage of reads with primers — in the box below, you’ll have to scroll all the way to the right to see those file names.
(An alternative way, of course, would just be to scroll through the entire output file, but this is (even) more of a hassle, as Cutadapt has quite some output.)
grep -B7 "with adapter:" results/cutadapt/logs/slurm-cutadapt.outCommand line parameters: -a GTGTGYCAGCMGCCGCGGTAA...ATTAGAWACCCBNGTAGTCC -A GGACTACNVGGGTWTCTAAT...TTACCGCGGCKGCTGRCACAC --trimmed-only --cores 8 --output results/cutadapt/NW102AB_R1.fastq.gz --paired-output results/cutadapt/NW102AB_R2.fastq.gz data/fastq/NW102AB_R1.fastq.gz data/fastq/NW102AB_R2.fastq.gz
Processing paired-end reads on 8 cores ...
Finished in 0.875 s (68.148 µs/read; 0.88 M reads/minute).
=== Summary ===
Total read pairs processed: 12,844
Read 1 with adapter: 12,798 (99.6%)
Read 2 with adapter: 12,541 (97.6%)
--
Command line parameters: -a GTGTGYCAGCMGCCGCGGTAA...ATTAGAWACCCBNGTAGTCC -A GGACTACNVGGGTWTCTAAT...TTACCGCGGCKGCTGRCACAC --trimmed-only --cores 8 --output results/cutadapt/NW102C_R1.fastq.gz --paired-output results/cutadapt/NW102C_R2.fastq.gz data/fastq/NW102C_R1.fastq.gz data/fastq/NW102C_R2.fastq.gz
Processing paired-end reads on 8 cores ...
Finished in 0.801 s (55.030 µs/read; 1.09 M reads/minute).
=== Summary ===
Total read pairs processed: 14,549
Read 1 with adapter: 14,499 (99.7%)
Read 2 with adapter: 14,211 (97.7%)
--
# [...output truncated...]Finally, let’s check the output dir, which contains the trimmed FASTQ files we’ll use in the next step of the workflow:
ls results/cutadaptNW102AB_R1.fastq.gz NW103C_R1.fastq.gz NW203A_R1.fastq.gz NW304BC_R1.fastq.gz NW403A_R1.fastq.gz NW404BC_R1.fastq.gz W103AB_R1.fastq.gz W204BC_R1.fastq.gz W303AB_R1.fastq.gz W304C_R1.fastq.gz W404A_R1.fastq.gz
NW102AB_R2.fastq.gz NW103C_R2.fastq.gz NW203A_R2.fastq.gz NW304BC_R2.fastq.gz NW403A_R2.fastq.gz NW404BC_R2.fastq.gz W103AB_R2.fastq.gz W204BC_R2.fastq.gz W303AB_R2.fastq.gz W304C_R2.fastq.gz W404A_R2.fastq.gz
NW102C_R1.fastq.gz NW201AB_R1.fastq.gz NW203BC_R1.fastq.gz NW305AB_R1.fastq.gz NW403BC_R1.fastq.gz W101AB_R1.fastq.gz W103C_R1.fastq.gz W205A_R1.fastq.gz W303C_R1.fastq.gz W403AB_R1.fastq.gz W404BC_R1.fastq.gz
NW102C_R2.fastq.gz NW201AB_R2.fastq.gz NW203BC_R2.fastq.gz NW305AB_R2.fastq.gz NW403BC_R2.fastq.gz W101AB_R2.fastq.gz W103C_R2.fastq.gz W205A_R2.fastq.gz W303C_R2.fastq.gz W403AB_R2.fastq.gz W404BC_R2.fastq.gz
NW103AB_R1.fastq.gz NW201C_R1.fastq.gz NW304A_R1.fastq.gz NW305C_R1.fastq.gz NW404A_R1.fastq.gz W101C_R1.fastq.gz W204A_R1.fastq.gz W205BC_R1.fastq.gz W304AB_R1.fastq.gz W403C_R1.fastq.gz
NW103AB_R2.fastq.gz NW201C_R2.fastq.gz NW304A_R2.fastq.gz NW305C_R2.fastq.gz NW404A_R2.fastq.gz W101C_R2.fastq.gz W204A_R2.fastq.gz W205BC_R2.fastq.gz W304AB_R2.fastq.gz W403C_R2.fastq.gzls -lh results/cutadapt-rw-rw----+ 1 jelmer PAS0471 1.9M Mar 6 14:46 NW102AB_R1.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 2.4M Mar 6 14:46 NW102AB_R2.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 2.2M Mar 6 14:46 NW102C_R1.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 2.8M Mar 6 14:46 NW102C_R2.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 1.8M Mar 6 14:46 NW103AB_R1.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 2.4M Mar 6 14:46 NW103AB_R2.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 2.2M Mar 6 14:46 NW103C_R1.fastq.gz
-rw-rw----+ 1 jelmer PAS0471 2.9M Mar 6 14:46 NW103C_R2.fastq.gz
# [...output truncated...]6 Bonus content
6.1 The FASTQ format
FASTQ is a very common output format of high-throughput sequencing machines. Like most genomic data files, these are plain text files. Each sequence that is read by the sequencer (i.e., each “read”) forms one FASTQ entry represented by four lines. The lines contain, respectively:
- A header that starts with
@and e.g. uniquely identifies the read - The sequence itself
- A
+(plus sign) - One-character quality scores for each base in the sequence
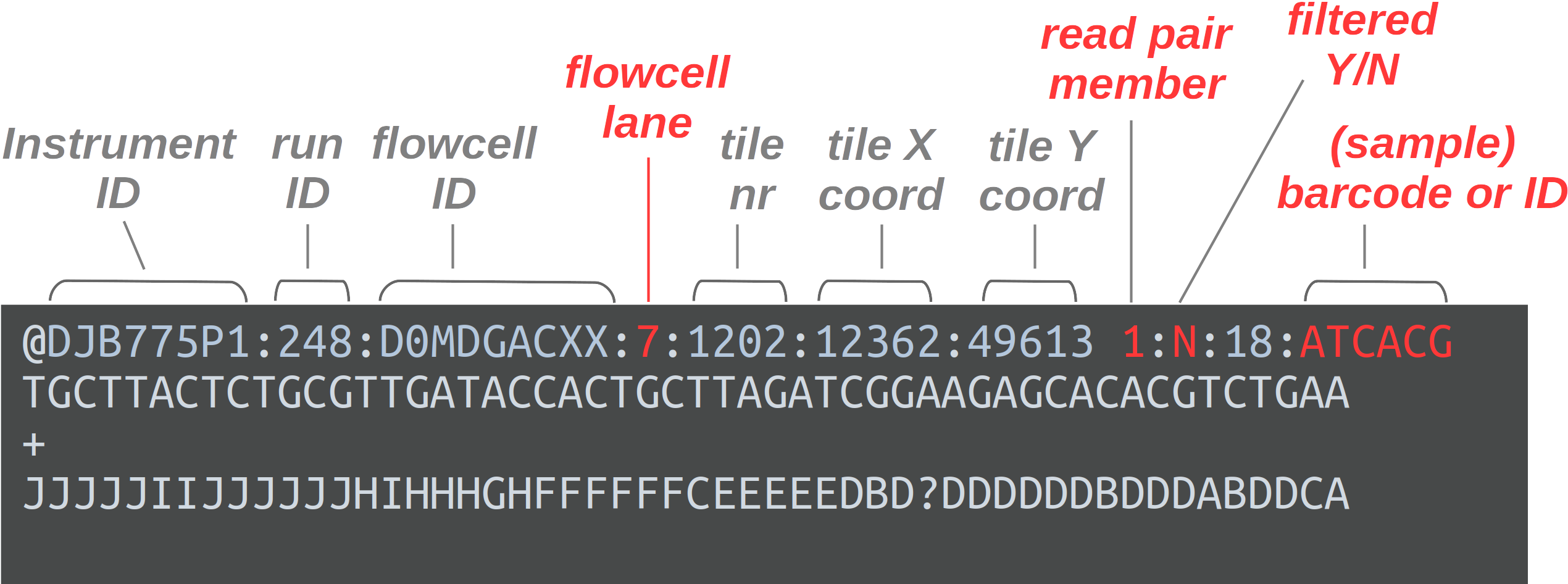
The header line is annotated, with some of the more useful components highlighted in red.
For viewing purposes, this read (at only 56 bp) is shorter than what is typical.
The “Q” in FASTQ stands for “quality”, to contrast this format with FASTA, a more basic and generic sequence data format that does not include base quality scores. FASTQ files have the extension .fastq or .fq, but they are very commonly gzip-compressed, in which case their name ends in .fastq.gz or .fq.gz.
The quality scores we saw in the read above represent an estimate of the error probability of the base call. Specifically, they correspond to a numeric “Phred” quality score (Q), which is a function of the estimated probability that a base call is erroneous (P):
Q = -10 * log10(P)
For some specific probabilities and their rough qualitative interpretations for Illumina data:
| Phred quality score | Error probability | Rough interpretation | ASCII character |
|---|---|---|---|
| 10 | 1 in 10 | terrible | + |
| 20 | 1 in 100 | bad | 5 |
| 30 | 1 in 1,000 | good | ? |
| 40 | 1 in 10,000 | excellent | ? |
This numeric quality score is represented in FASTQ files not by the number itself, but by a corresponding “ASCII character” (last column in the table). This allows for a single-character representation of each possible score — as a consequence, each quality score character can conveniently correspond to (& line up with) a base character in the read. (For your reference, here is a complete lookup table — look at the top table, “BASE=33”).
6.2 Viewing FASTQ files
Next, we’ll take a peak inside one of these FASTQ files.
The head command prints the first lines of a file. Let’s use it try to and print 8 lines, which should show us two reads:
head -n 8 data/fastq/NW102AB_R1.fastq.gz�
Խے�8�E��_1f�"�QD�J��D�fs{����Yk����d��*��
|��x���l�j�N������?������ٔ�bUs�Ng�Ǭ���i;_��������������|<�v����3��������|���ۧ��3ĐHyƕ�bIΟD�%����Sr#~��7��ν��1y�Ai,4
w\]"b�#Q����8��+[e�3d�4H���̒�l�9LVMX��U*�M����_?���\["��7�s\<_���:�$���N��v�}^����sw�|�n;<�<�oP����
i��k��q�ְ(G�ϫ��L�^��=��<���K��j�_/�[ۭV�ns:��U��G�z�ݎ�j����&��~�F��٤ZN�'��r2z}�f\#��:�9$�����H�݂�"�@M����H�C�
�0�pp���1�O��I�H�P됄�.Ȣe��Q�>���
�'�;@D8���#��St�7k�g��|�A䉻���_���d�_c������a\�|�_�mn�]�9N������l�٢ZN�c�9u�����n��n�`��
"gͺ�
���H�?2@�FC�S$n���Ԓh� nԙj��望��f �?N@�CzUlT�&�h�Pt!�r|��9~)���e�A�77�h{��~�� ��
# [...output truncated...]Ouch! 😳 What went wrong here? (Click for the solution)
What happened here is that we are directly seeing the contents of the compressed file, which is simply not human-readable.To get around the problem we just encountered with head, we might be inclined to uncompress these files, which we could do with the gunzip command. However, uncompressed files take up several times as much disk storage space as compressed ones. Fortunately, we don’t need to decompress them:
- Almost any bioinformatics tool will accept compressed FASTQ files.
- We can still view these files in compressed form, as shown below.
Instead, we’ll use the less command, which will automatically display gzip-compressed files in human-readable form:
less -S data/fastq/NW102AB_R1.fastq.gz@M02815:77:000000000-KPK85:1:2101:3678:10660 1:N:0:CCTAAGAC+TTCTAGCT
CGAGCAATCCACTCGAGTGCCAGCAGCCGCAGTAATACGGAGGGTGCGAGCGTTGTCCGGAATCACTGGGCGTAAAGGGCGCGTAGGCGGCGCGGATAGTCGGCGGTGAAAGCCCGGAGCTCAACTCCGGGTCGGCCGTCGATACTTCCGGGCTTGAGCACTGTAGAGGCAGATGGAATTCCGGGTGTAGCGGTGGAATGCGTAGAGATCCGGAAGAACACCGGTGGCGAAGGCGGTCTGCTGGGCAGTTGCTGACGCTGATGCGCGACAGCGTGGGGAGCAAACAGGATTAGATACC
+
CCCCCGGGGGGGGGGGGGGFGGGGGGGGGG+CFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGEGGGGGGGGGGGGGGGGGGGGDGGGGGGGGGGGGGGFGGFGFFFFEBFFGFFFDGFGFGBFGFGFGFFFF6?FFFGBF?FBFFF
@M02815:77:000000000-KPK85:1:2108:2535:14400 1:N:0:CCTAAGAC+TTCTAGCT
CGAGCAATCCACTCGAGTGTCAGCCGCCGCGGTAATACAGAGGTCCCGAGCGTTGTTCGGATTCATTGGGCGTAAAGGGTGCGTAGGCGGCGGGGAAAGTCTGATGTGAAATCCTGGGGCTCAACCCTGGAACTGCATTGGATACTTCCTTGCTAGAGTACTGGAGAGGAAACTGGAATTTACGGTGTAGCAGTGAAATGCGTAGAGATCGTAAGGAAGACCAGTGGCGAAGGCGAGTTTCTGGACAGTTACTGACGCTGAGGCACGAAGGCCAGGGGAGCAAACGGGATTAGATACC
+
CCCCCCGFGFGGGC-FFFGFGFFGGDFFGGGGGECGEGGAEGGGGGGGFGGDGG7CFFGGDCCFGGFCF8FGGGGGGCEGDGGGGGCGGGGGGDEGGGGBFGGDFGGGDG<DFGGGGCEGGGD:FFGGGGFFGFGGFFFFGGGFGGCFGGFGGGGG9CGCGGGG7FGGC:FFGGGGGFGG<?FCGGGGGGGGGGG9CG<ACC?EG5CFGGGGF8CCCC:C@FGCFGGGGGC58=EEG8??77:9@:<3A>7AGFGGGGC?DFC?5<5>>BGGGFGGGGG>4?C42::3:DG=><<*)*less -S suppresses line-wrapping: lines in the file will not be “wrapped” across multiple lines
Exercise: Explore the file with less
less doesn’t print stuff to screen but instead opens it in a “pager”. After running the command above, you should be viewing the file inside the less pager.
You can move around in the file in several ways: by scrolling with your mouse, with up and down arrows, or, if you have them, PgUp and PgDn keys (also, u will move up half a page and d down half a page).
Notice you won’t get your shell prompt back until you press q to quit less.
Footnotes
For a full list of installed software at OSC: https://www.osc.edu/resources/available_software/software_list↩︎
Attribution: Some of the FastQC example plots were taken from here.↩︎
Checks for adapters at the ends of reads. Since we have to remove primers anyway, any adapters past the primers will be automatically removed.↩︎
Will throw a Fail but this is not meaningful here: metabarcoding data has many duplicate sequences by design.↩︎
As with the previous module, this will throw a Fail but this is not meaningful here: metabarcoding data has many duplicate sequences by design.↩︎
Useful in a whole-genome sequencing context or to detect contamination.↩︎
Therefore, our FastQC analysis will run sequentially (1-by-1) for each file, not in parallel.↩︎
Using the shell language Bash, specifically↩︎
And very large jobs can sometimes take hours to start, but our jobs are small so that should not happen.↩︎
This is called “command substitution”.↩︎
This is not the default: Cutadapt is even more commonly used to remove adapters, and then this doesn’t apply↩︎