High-throughput sequencing and genomes
2026-01-29
Sanger sequencing
- Sanger sequencing almost always starts with PCR amplification of the target DNA region —
as illustrated by Dr. Popp last week:
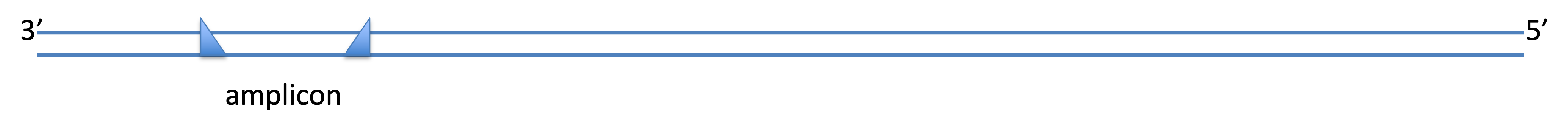
Therefore, to design primers, you must know something about the target sequence in advance — this can be highly limiting
The sequenced fragment can be up to about 800-1,000 bp
Sanger sequencing
Sequencing itself is performed by synthesizing a new DNA strand with fluorescently-labeled nucleotides, using a different color for each base (
A,C,G,T)The final result is a chromatogram that can be “base-called”:
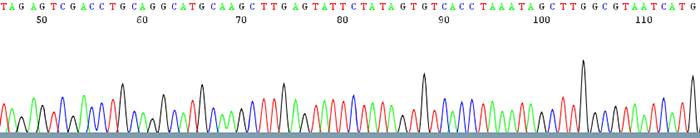
The entire human genome was sequenced with Sanger technology!
How many basepairs is that? Want to guess how much it cost to do this?
Sequencing cost through time
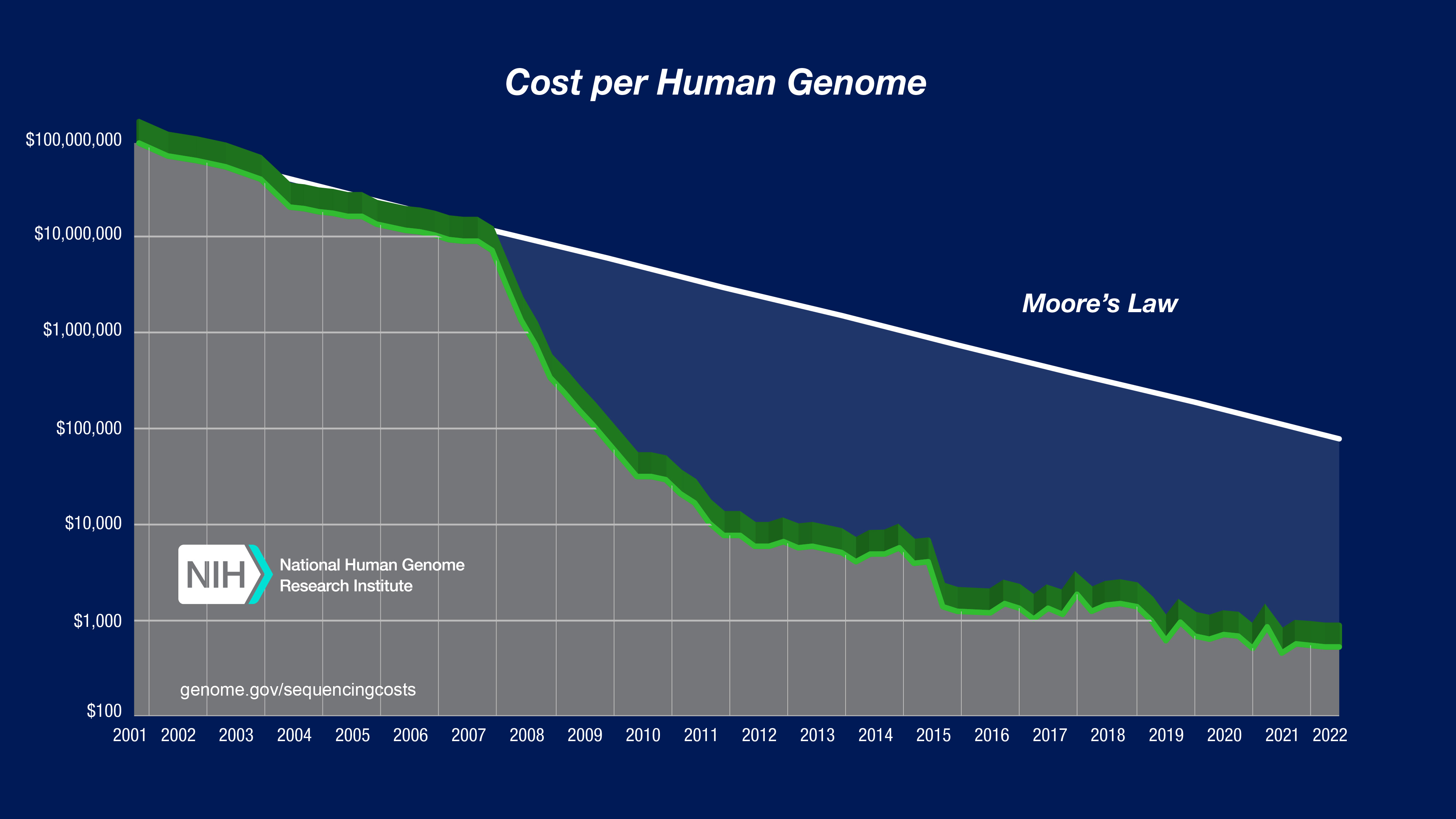

Image generated by Adobe Firefly
Omics
Let’s start with the big picture – HTS data underlies several of these main “omics” approaches:
Copyright ThermoFisher
Error rates
A read’s sequence may differ from the actual DNA sequence it originated from:
- The read can have base-calling errors, missing bases, or extra bases
- When the base calling software is not confident, it can also return
Ns (= undetermined)
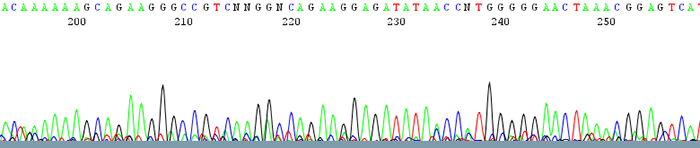
When you receive HTS reads, base calls have typically been made already.
Every base call is accompanied by a quality score, representing the estimated error probability.
Correcting sequencing errors
To overcome sequencing errors, every base can be sequenced multiple times –
i.e., obtaining a “depth of coverage” greater than 1:
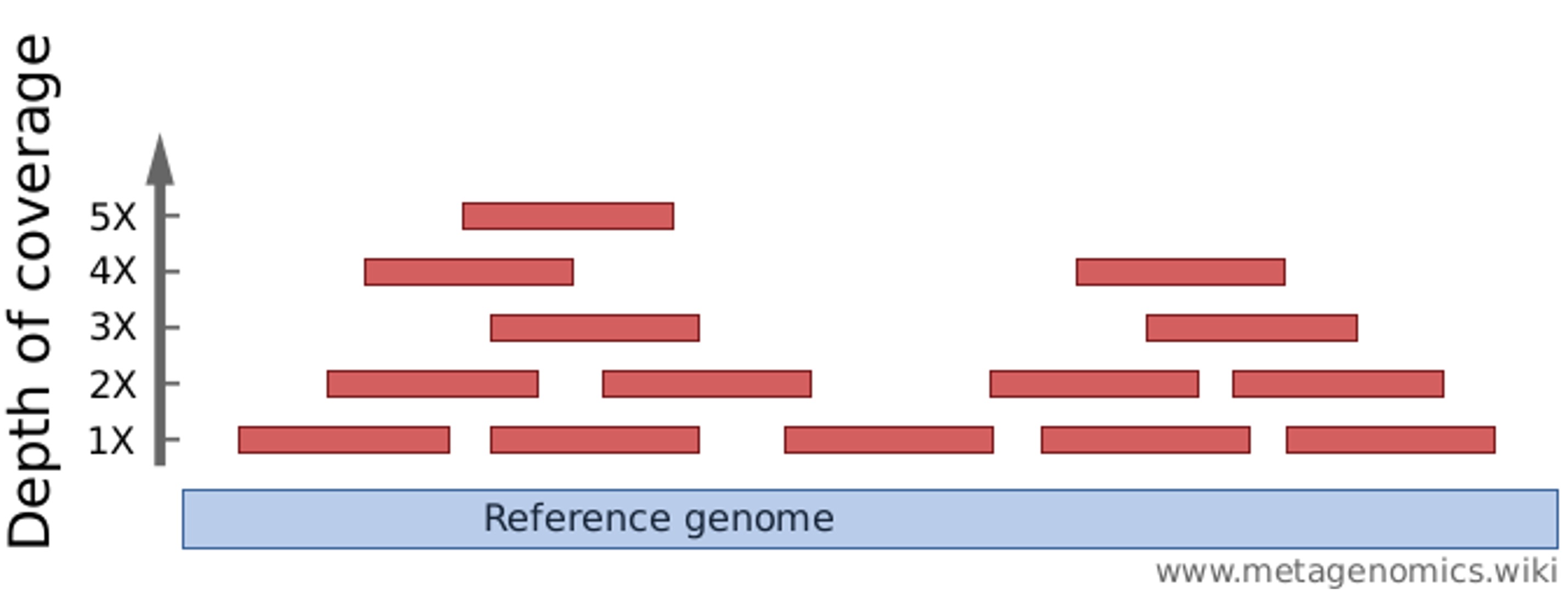
Which natural phenomenon might complicate this effort?
Genetic variation among and (for diploid organisms) within individualsTypical depths of coverage: ~50-100x for genome assembly & 10-30x for variant typing (!)
Libraries and library prep
- In a HTS context, a “library” is a collection of DNA fragments ready for sequencing
- These fragments can number in the millions or billions and are often randomly generated from input like genomic DNA:
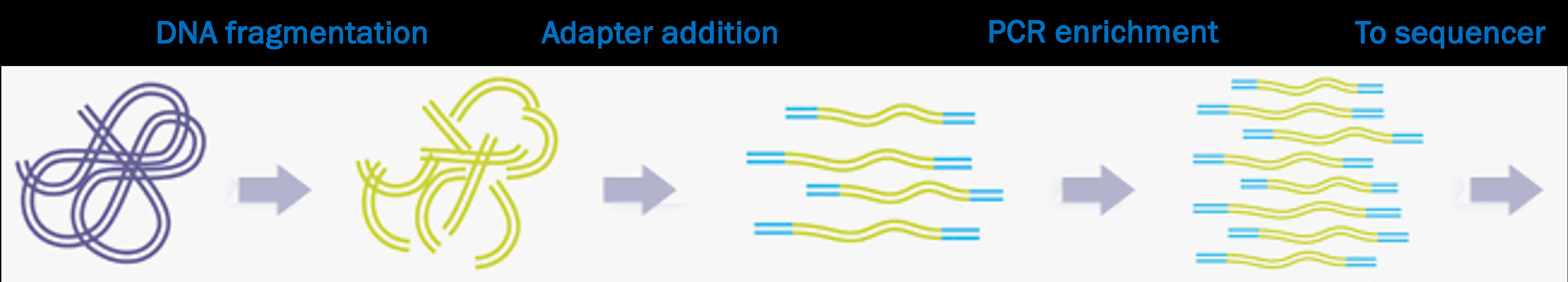
An overview of the library prep procedure. This is typically done for you by a sequencing facility or company.
Libraries and library prep
- After library prep, each DNA fragment is flanked by several types of short sequences that together make up the “adapters”:

Multiplexing!
Adapters can include “indices” or “barcodes” to identify individual samples, so many samples can be combined (multiplexed) into a single library
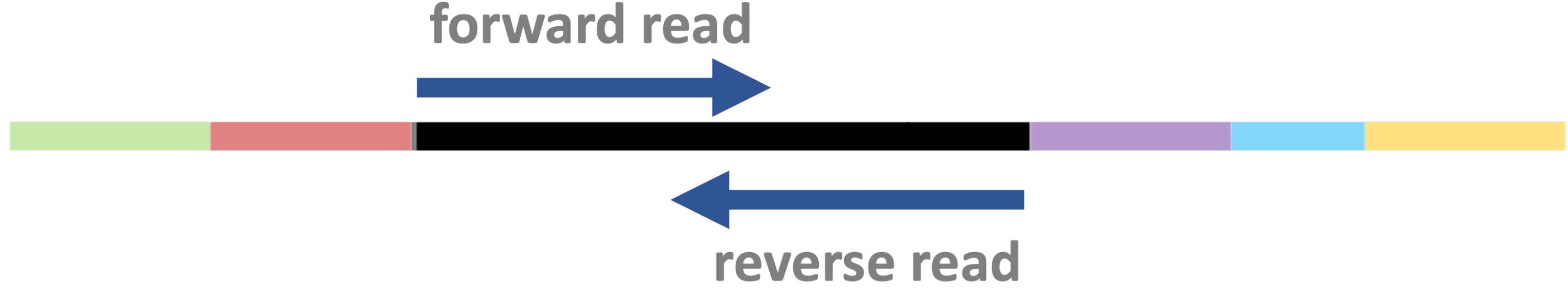
Paired-end vs. single-end sequencing
- Fragments can be sequenced from both ends as shown below —
this is called “paired-end” (PE) sequencing:

- When sequencing is instead single-end (SE), no reverse read is produced:

Fragment size variation
- DNA fragment size varies – by design and because of limited precision in size selection
What happens when a fragment is shorter than the length of a single F or R read?
“Adapter read-through”: the final bases in the resulting reads will consist of adapter sequence (these should be removed before downstream analysis)

Fragment size variation
- DNA fragment size varies – by design and because of limited precision in size selection
What happens when a fragment is shorter than the combined F + R read length?
Overlapping reads (this can be useful!):
How Illumina sequencing works
We start with fragments that have been attached to a flow cell —
this image shows just two, but millions are present simultaneously

How Illumina sequencing works
Sequencing is performed by synthesizing a new strand using fluorescently-labeled bases,
and taking a picture each time a new nucleotide is incorporated:

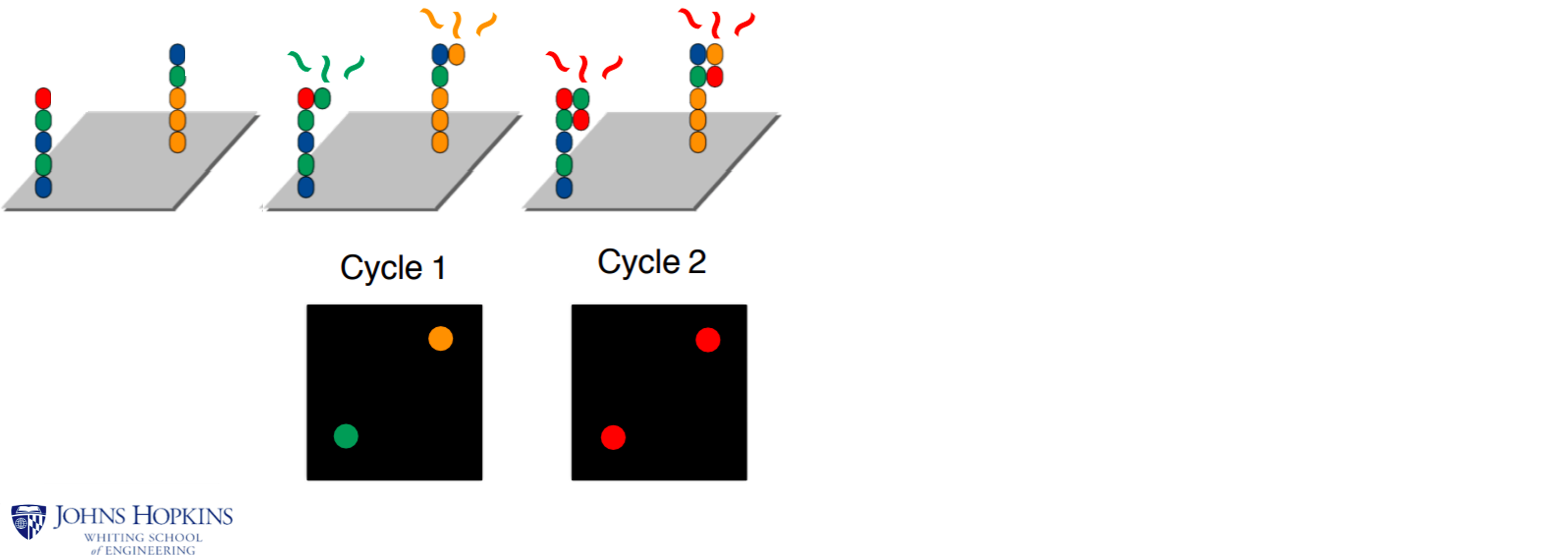
How Illumina sequencing works
Sequencing is performed by synthesizing a new strand using fluorescently-labeled bases,
and taking a picture each time a new nucleotide is incorporated:
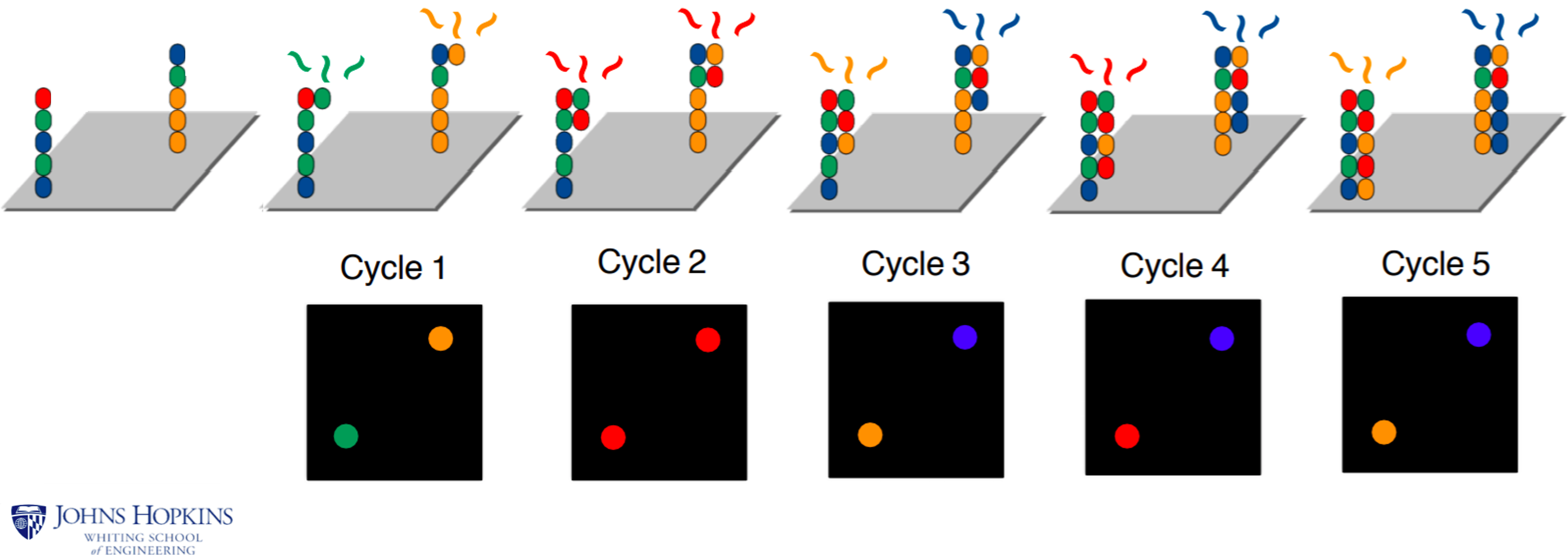
How Illumina sequencing works
Sequencing is performed by synthesizing a new strand using fluorescently-labeled bases,
and taking a picture each time a new nucleotide is incorporated:
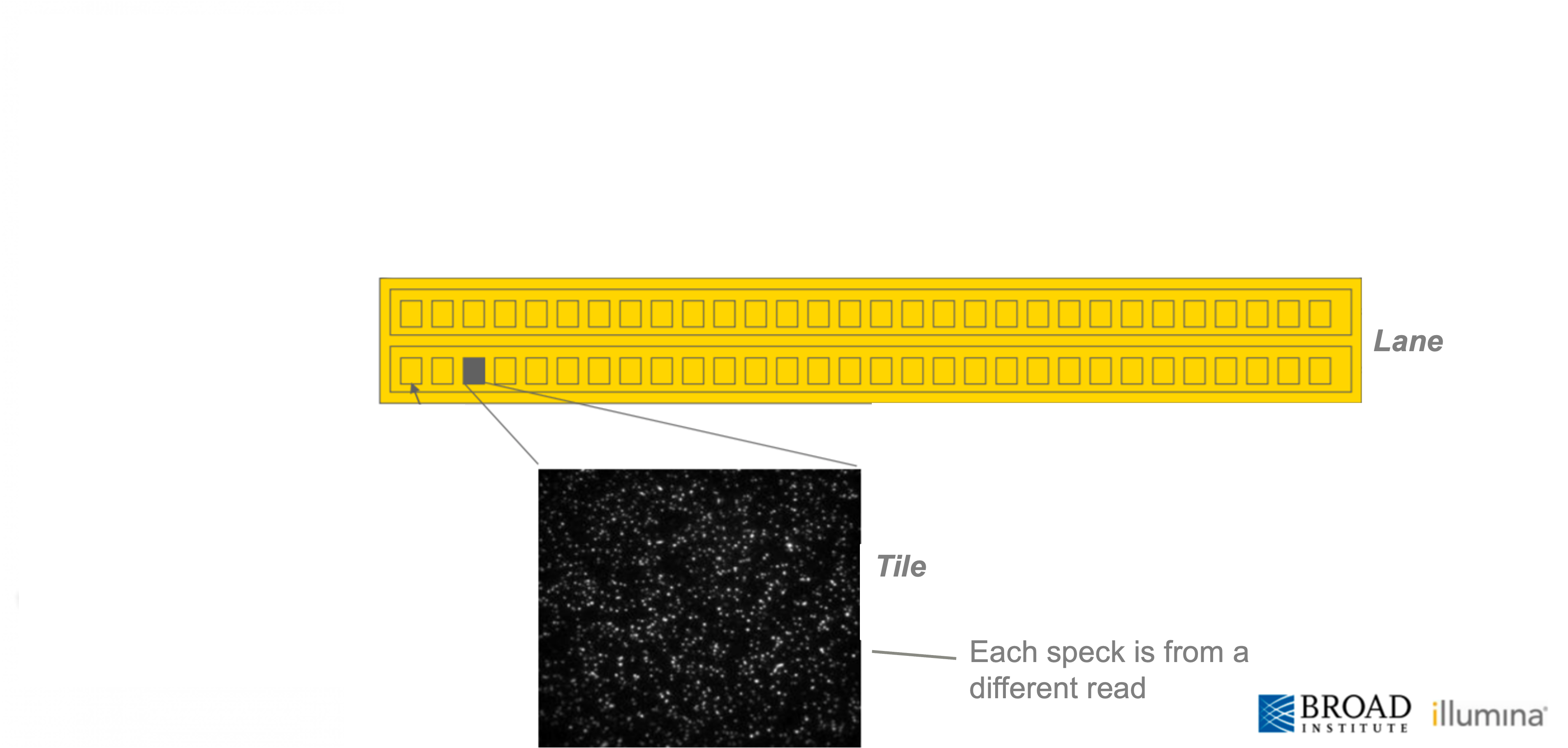
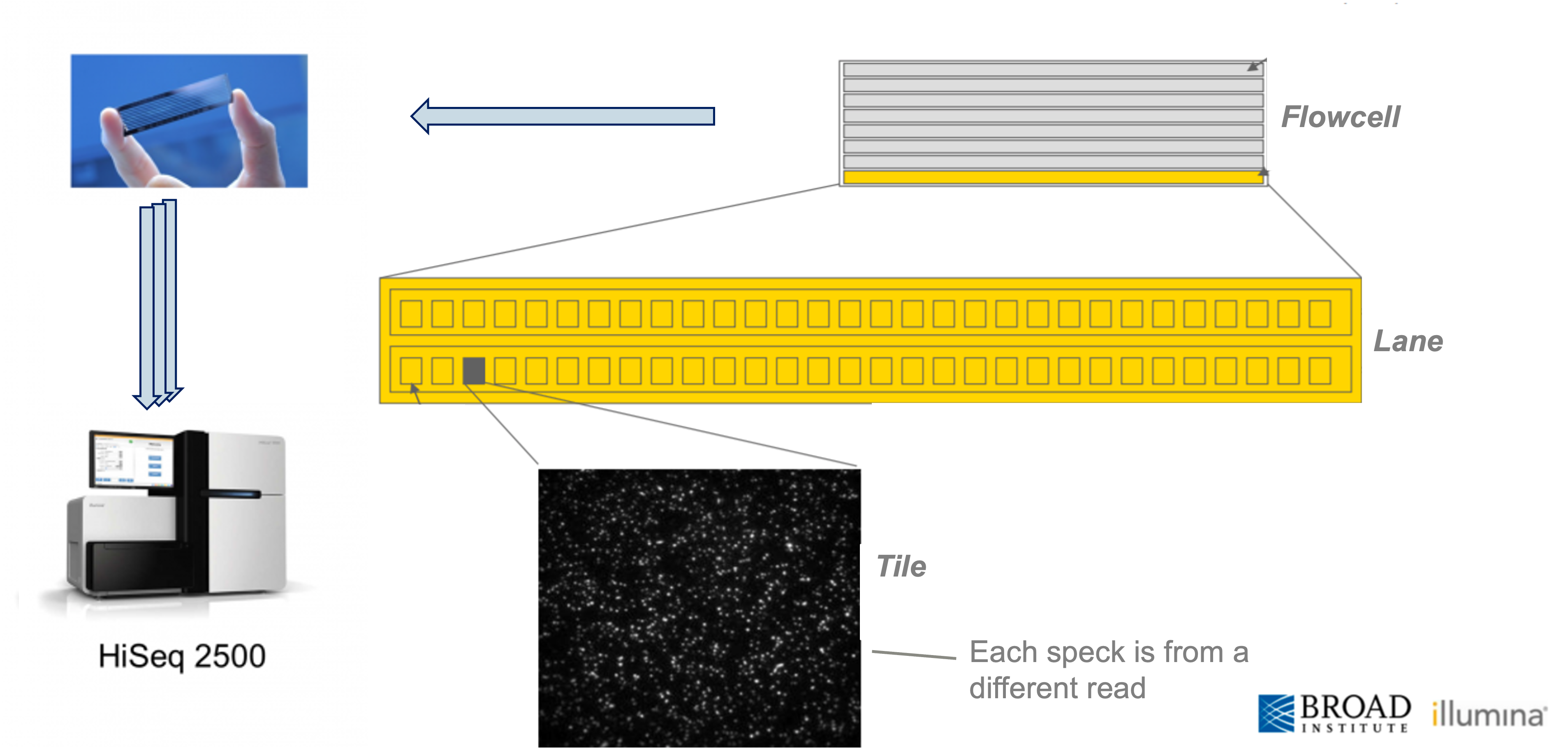
The scale of Illumina sequencing

The scale of Illumina sequencing
The scale of Illumina sequencing
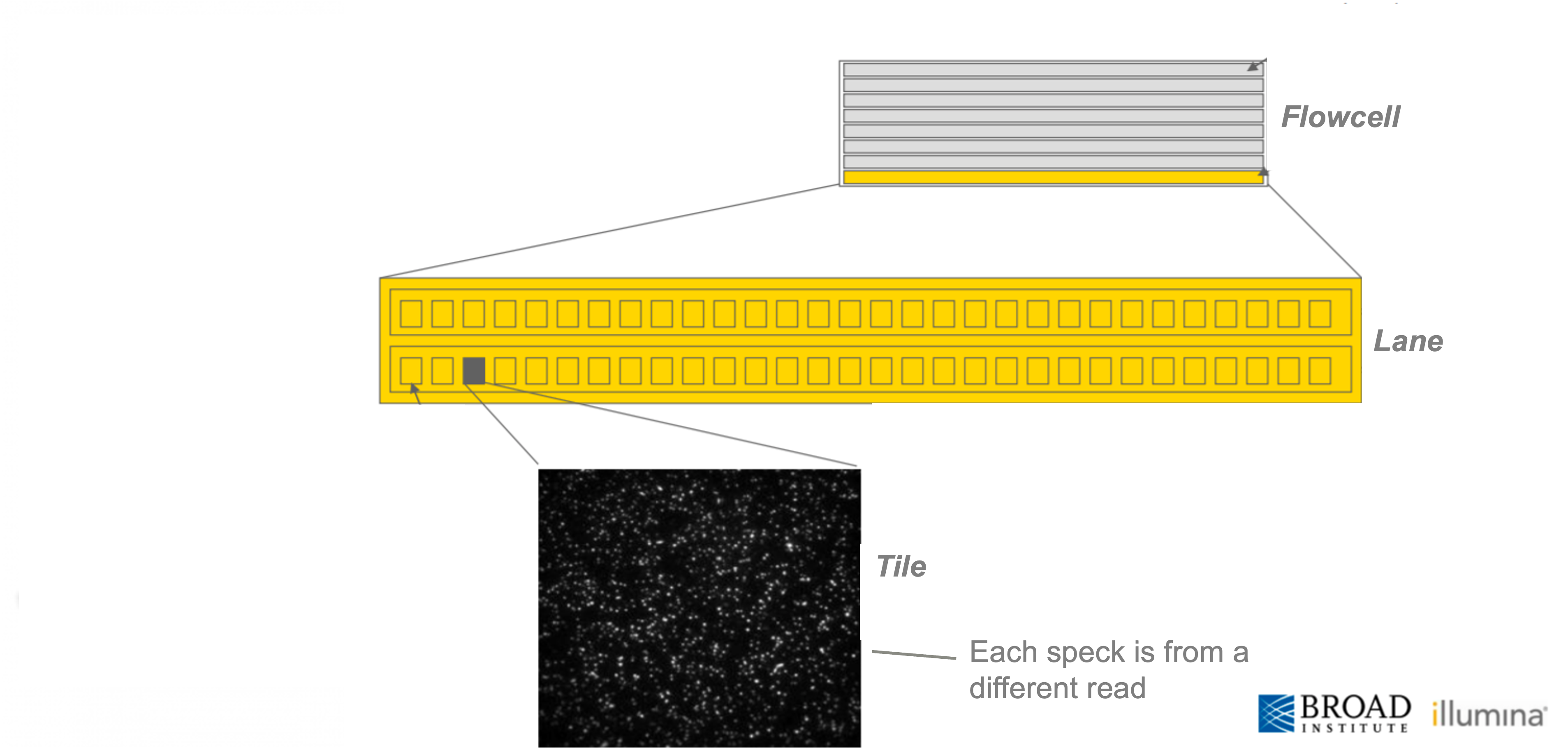
The scale of Illumina sequencing
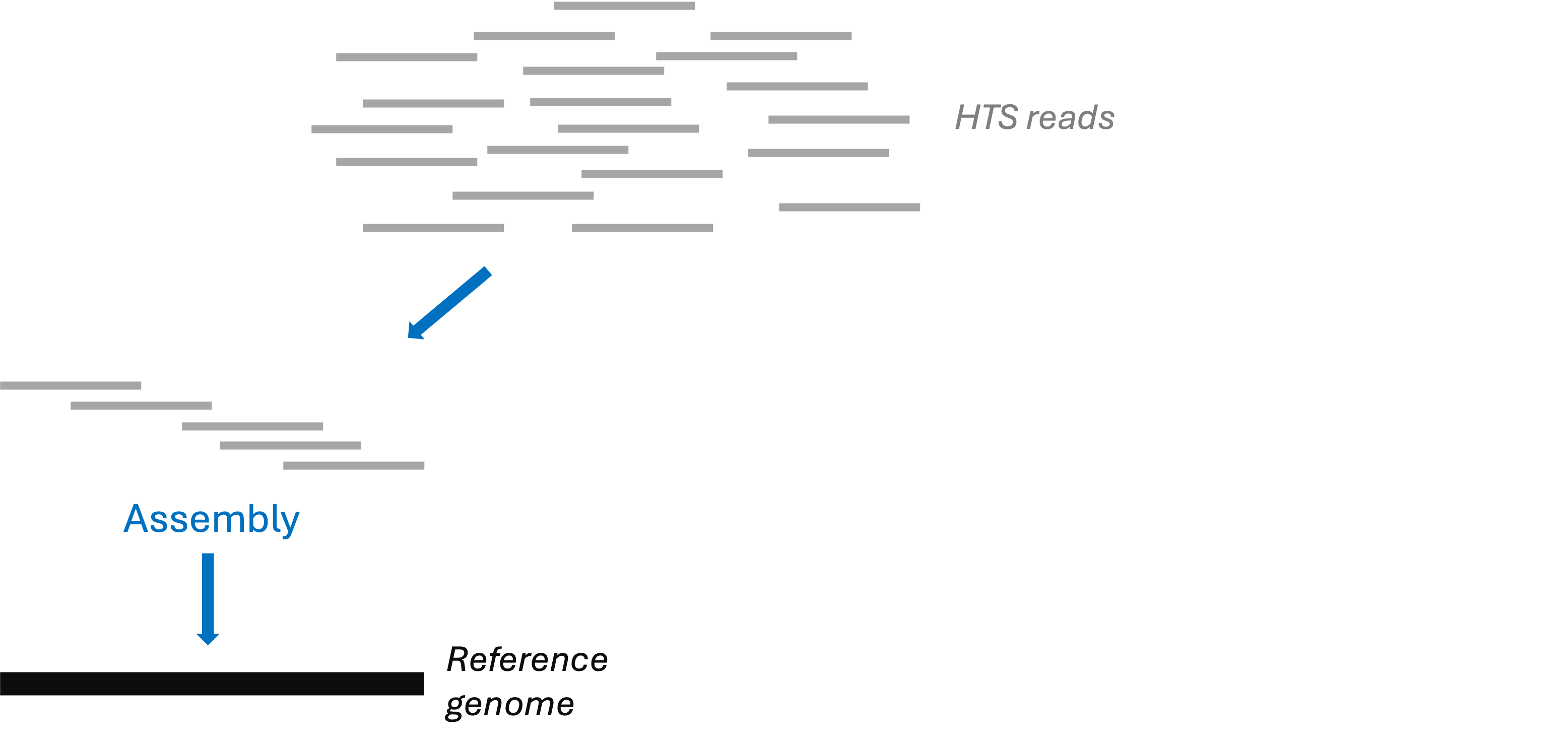
Reference genomes
Many HTS applications either require a “reference genome” or involve its production:
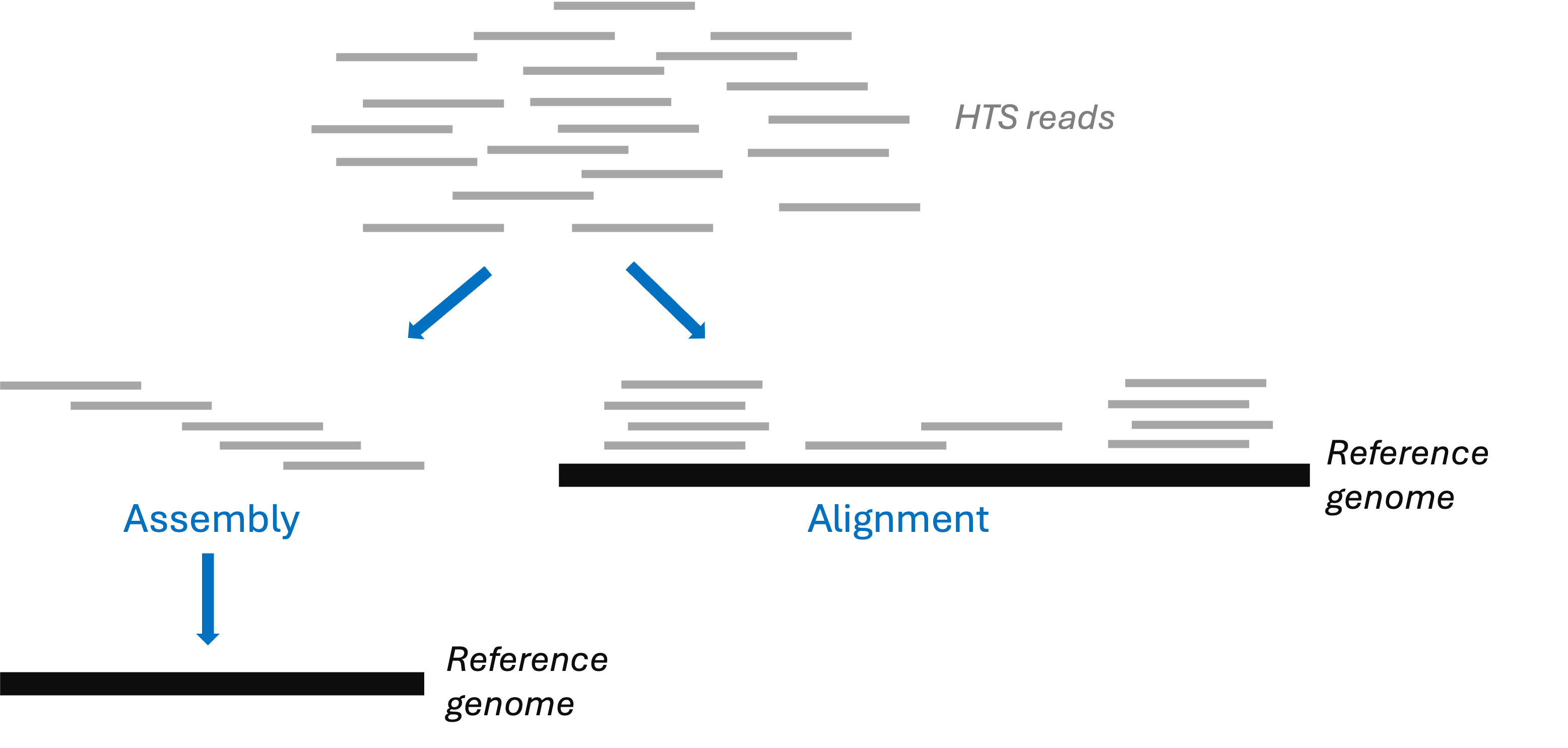
Reference genomes
Many HTS applications either require a “reference genome” or involve its production:
There is enormous variation in genome size

And enormous growth of genomes in databases
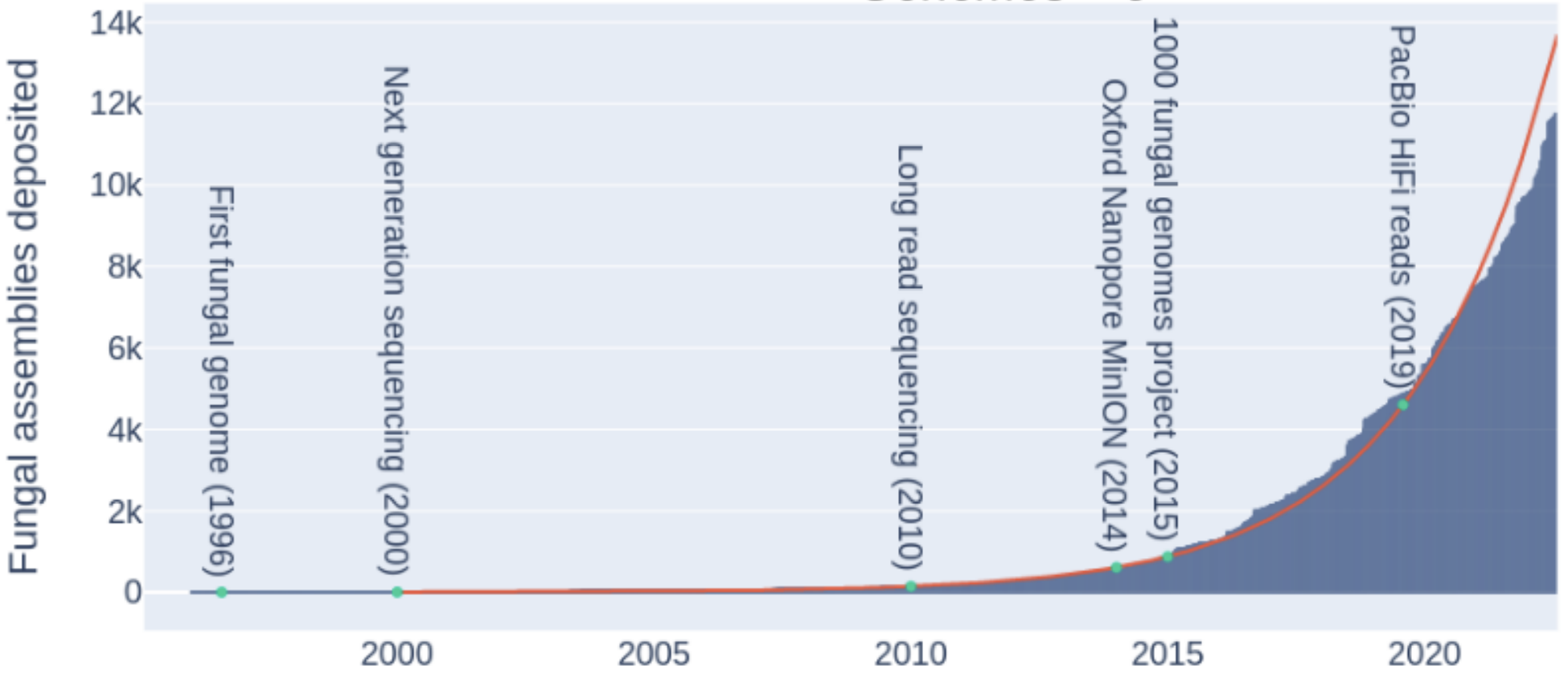
Konkel and Slot (2023)
The Garrigós et al. 2025 dataset
The labs this and next week are organized around the data set from Garrigós et al. (2025):

This paper uses paired-end Illumina RNA-Seq data to study gene expression in Culex pipiens mosquitos infected with two different malaria-causing Plasmodium protozoans.
